

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Neuroblastoma (NB) is the most common malignant disease in children and accounts for approximately 15% of all childhood cancer death [1]. NB is an extracranial solid tumor originating from multipotent neural crest stem cells [2,3]. Significant similarities in gene expression profiles have been detected between normal neuroblasts and malignant NB [4], thereby supporting the notion that malignant NB resemble neural stem cells. Unfavorable NB is poorly differentiated [5,6].
Current treatment of NB consists of chemotherapy, surgery, and radiation [7]. For many years, it has been recognized that the exposure of NB cells to retinoids (all-trans-RA, 13-cis-RA, and 9-cis-RA) results in reduced tumor cell proliferation and cell differentiation [8]. Retinoids are the biologically active products from provitamin A carotenoids including α-carotene, β-carotene, and β-cryptoxanthin [9]. Central cleavage by carotene-15,15'-oxygenase is a major pathway leading to vitamin A production from provitamin A carotenoids in the body [10]. Retinoic acid (RA) is the biologically active form of vitamin A-that is, retinol, retinal, and retinyl esters, which are precursors of RA. RA exerts its biological role in embryonic development and organogenesis, cell proliferation, differentiation, and apoptosis through its signaling mediated by the activation of specific nuclear receptors [10,11]. Comprehensive studies in vivo and in vitro have provided the tumor suppressor functions of vitamin A through its metabolites. Retinoids have been employed in clinical trials in children with advanced NB. A series of randomized clinical trials have shown the benefit of a combination of RA with chemotherapy in acute promyelocytic leukemia during induction therapy [12,13]. There is no clear roadmap for prioritizing retinoids or combination strategies for clinical evaluation. Nonetheless, decisions regarding the prioritization of clinical research opportunities must be made the basis of the best available information, including the results of animal studies and in vitro cell studies.
DLK1 (Drosophila delta-like 1 homologue) is a type I transmembrane protein belonging to the epidermal growth factor (EGF)-like homeotic gene family, and is homologous to the notch/delta/serrate family [14]. DLK1 is also known as preadipocyte factor 1 (pref-1), fetal antigen (FA1), pG2, and ZOG [14]. DLK1 is expressed extensively in immature cells and down-regulated during fetal development [15-17], thereby suggesting that DLK1 performs an important role in stem/progenitor cells. DLK1 is involved in the differentiation of several cell types, including preadipocytes [18], hematopoietic stem cells [19,20] and lymphoid progenitors [21,22]. DLK1 expression has also been detected in a variety of tumor cells, including NB [23, 24] gliomas [25], small-cell lung carcinoma [24], and leukemia [19,20]. In a previous study conducted by this researcher, DLK1 was reported to be expressed preferentially in poorly differentiated NB cells, and DLK1 expression was reduced when cells were induced to differentiate by RA [26]. Most importantly, the results of previous studies have shown that DLK1 may enhance cancer cell stemness. Evidence also appears to suggest that DLK1 may inhibit tumor cell differentiation and increase tumorigenic potential [19,25], although the mechanisms underlying this effect remain unclear.
To the extent possible, it is crucial to determine whether the signaling pathways perform important functions in preclinical models, such as animal or in vitro cell lines. The mitogen-activated protein kinase (MAPK) signaling pathways are involved in a variety of cellular events, including growth, differentiation, and stress responses [27,28]. It has been previously reported that ERK (p42/44 MAPK) was phosphorylated in neuronal differentiation [28], and DLK1 was involved in multi-signaling pathways, including the ERK phosphorylation pathway [26]. Both PD98059 and U0126 have long been used as specific inhibitors of the ERK (p42/44 MAPK) pathway [29-31].
Based on the evidence discussed above, the principal hypothesis of this study was that RA and DLK1 perform important roles in the differentiation of neuronal cells. A combination of RA and DLK1 may induce more significant effects on cell differentiation, and thereby block NB tumor cell progression. This hypothesis represents a novel perspective on the NB cell differentiation induced by RA and DLK1 knockdown. This study has the potential not only to elucidate the roles of DLK1 and RA in the regulation of NB stem cell phenotype and differentiation, but also to provide mechanistic insights into the progression of malignant tumors. The results of this study will benefit the prevention and treatment of this deadly childhood cancer.
Materials and Methods
Cell culture and differentiation
SK-N-BE(2)C [BE(2)C] NB cells were cultured in Minimum Essential Medium and F12 (1:1) at 37℃ in an atmosphere of 5% CO2 and 95% air. The media were supplemented with 10% fetal bovine serum (v/v). For all experiments, BE(2)C cells were cultured to 60-80% confluence. The medium was replaced every other day. For viral infection, the cells were infected at 30-50% confluence as described previously [32]. In order to induce differentiation, BE(2)C cells were treated with various concentrations of all-trans-retinoic acid (RA). U0126 and PD98059 were purchased from Cell Signaling Technology Inc. (Danvers, MA).
RNA interference
The following siRNA oligonucleotides against human DLK1 were purchased from Dharmacon (Lafayette, CO): 5'-GCACC UGCGUGGAUGAUGAUU-3' for DLK1 siRNA (05) and 5'-GGACGAUGGCCUCUAUGAAUU-3' for DLK1 siRNA (07). The sequence of control siRNA was UAACAAUGAGAGCA CGGCUUU. Transfection with siRNAs was performed using DharmaFECT (Dharmacon, Lafayette, CO) according to the manufacturer's instruction.
Dr. Tadanori Mayumi (University of Tokyo, Tokyo, Japan) kindly provided the lentiviral vectors: pHM5-H1 and CS-CDF-EG-PRE-K1f, for the construction of short hairpin RNA (shRNA) [33]. Oligonucleotides encoding for both strands of DLK1 sequences were annealed and inserted into pHM5-H1 between BglII and XbaI sites: 5'- GATCCCC GGTGTCCATGAAAGA GCTC TTCAAGAGA GAGCTCTTTCATGGACACC TTTTT GGAAA T-3' (sense strand) and 5'- CTAGA TTTCC AAAAA GGTGTCCATGAAAGAGCTC TCTCTTGAA GAGCTCTTTC ATGGACACC GGG-3' (antisense strand) for ShRNA-DLK1. After sequence validation, the cassette containing the H1 promoter and a ShRNA-DLK1 sequence was inserted into CS-CDF-EG-PRE-K1f between EcoRI and Pst1 sites. The sequence was verified further by DNA sequencing. The empty lentiviral vector was employed as a negative control.
Western blotting
Cell lysates were prepared on ice in RIPA lysis buffer containing 50 mM Tris-Cl at pH 7.5, 150 mM NaCl, 1% Nonidet p-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1-2 mM PMSF, 1mM NaVO4, and 1mM NaF. The protein concentrations were measured using the BCA assay reagents (BioRad). 100 µg of whole cell lysate proteins were used for 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the separated proteins were transferred to a PVDF membrane. After blocking by incubation with 5% BSA in Tris-buffered saline for 1 hr at room temperature, the membrane was incubated using polyclonal antibody against rabbit phospho-extracellular signal-regulated kinase (ERK) (Cell Signaling Technology Inc, Danvers, MA). Horse-radish peroxidase-conjugated anti-IgG antibody was employed as the secondary antibody to detect protein bands to which the primary antibodies were bound by enhanced chemiluminescence. Anti β-actin (Sigma, St. Louis, MO) was used for a loading control.
Immunofluorescence
BE(2)C Cells were washed with PBS and fixed for 10 min with -20° C methanol at room temperature, then washed 3 times with PBS. The fixation was removed by aspiration and the cells were washed 3 times with PBS for 5 min each. The reaction was blocked with 10% horse serum in PBS for 1 hr at room temperature. Immunocytochemical analysis was conducted by incubating the cells with a 1:50 dilution of anti-DLK1 (R&D Systems, Minneapolis, MN) or anti-β-tubulin III (1:70, Sigma, St. Louis, MO) in 1% BSA for 2 hrs at room temperature. Primary antibody binding was detected with Alexa 594-conjugated donkey-anti-mouse (1:500, Invitrogen, Carlsbad, CA) or Alexa 488-conjugated goat-anti-rabbit (1:500, Invitrogen, Carlsbad, CA) for 1 hr at room temperature. Fluorescence microscopy was performed for image detection.
Results
RA induces neuronal differentiation in NB
Immunofluorescence was employed to evaluate the neuronal differentiation induced by RA in NB. RA markedly increased neurite growth, a hallmark of neuronal differentiation even at a concentration of 10 nM. The neuron-specific marker, β-Tubulin III, clearly demonstrated longer neurite growth in the RA-treated cells as compared to the non-treated cells. Neurite growth was increased in a concentration-dependent manner (Fig. 1A). Further, RA reduced the expression of some of the stem cell markers, including CD133, SOX2, and DLK1 (Fig. 1B). These observations corroborate the identification of RA as a true differentiation reagent for NB, and DLK1 is a bona fide stem cell/progenitor cell marker for immature NB cells.
Neuronal differentiation was increased with a combination of RA and DLK1 knockdown
To evaluate the effect of a combination therapy of RA and DLK1 knockdown on NB cell differentiation, the cells were transfected with two types of SiRNA DLK1 [SiRNA (05) or SiRNA (07)] oligonucleotides against human DLK1. The cells were then treated for 3 d with 10 nM RA. Both constructs inhibited the expression of endogenous DLK1 in BE(2)C cells (Fig. 2B). Although DLK1 knockdown spontaneously developed neuronal processes, neurite growth was increased dramatically by a combination with low concentration of RA treatment as revealed by immunofluorescence of the neuron-specific β-tubulin III (Fig. 2A). These results clearly indicate that the combination of RA and DLK1 knockdown has a more profound chemotherapeutic effect due to an induced increase in NB cell differentiation.
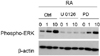
MAPK inhibitors block ERK phosphorylation induced by RA
In order to verify the involvement of the MAPK pathway in RA-induced NB cell differentiation, the MAPK specific inhibitors, U0126 and PD 98059, were administered. 5 µM of U0126 completely blocked ERK phosphorylation even at an RA treatment concentration of 10 µM. However, PD98059 partially blocked ERK phosphorylation at 1 µM or 10 µM of RA (Fig. 3).
The MAPK inhibitors, U0126 or PD98059, block RA-induced differentiation
To examine the effects of MAPK inhibitors on neuronal differentiation, RA was applied to BE(2)C cells at different concentrations (0 nM, 1 nM or 10 nM). Using specific inhibitors (PD98059 and U0126) of the ERK (p42/44 MAPK) pathway, unexpected differential effects of these inhibitors on RA-induced neuronal differentiation were observed. This neurite growth was completely blocked by U0126 but blocked only partially by PD98059 (Fig. 4). This result is consistent with the observed inhibition of ERK phophorylation as shown in Fig. 3. These findings indicate that the effects of these inhibitors are not exclusive, owing to an inhibition of the ERK MAPK pathway, and may raise some fundamental implications with regard to the inhibitors employed.
The MAPK inhibitors, U0126 or PD98059, block the neuronal differentiation induced by DLK1 knockdown
To evaluate the effect of MAPK inhibitors on the neuronal differentiation induced by DLK1 knockdown, BE(2)C cells were transfected with ShRNA-DLK1, and the cells were treated with various concentrations of U0126 or PD98059. As was observed under RA treatment (Fig. 4), the cells were differentiated by DLK1 knockdown; however, this differentiation was completely blocked by U0126 at all concentrations tested. On the other hand, PD98059 partially blocked the neuronal cell differentiation induced by DLK1 knockdown (Fig. 5). In order to confirm this finding, the cells differentiated by DLK1 knockdown were treated with each inhibitor for 3 days, after which the inhibitors were withdrawn via exchange with fresh media containing no inhibitors. After 2 days of withdrawal of the inhibitor, the neuronal differentiation of BE(2)C cells was fully recovered (Fig. 6) in each of the inhibitor-treated group. Even though U0126 blocked neuronal differentiation more dramatically than did PD98059, neurite growth was recovered in both inhibitor groups after the withdrawal of each inhibitor. These results demonstrated that the differentiation induced by RA and DLK1 knockdown may occur via the same pathway. Additionally, U0126 and PD98059 exerted different effects on neuronal differentiation.
Discussion
The present study explored the role of RA and DLK1 knockdown on the NB cell differentiation. The results from this investigation demonstrated that the pretreatment of human malignant NB cells with a combination of RA and DLK1 knockdown induced cell differentiation, thereby controlling malignant tumor growth through ERK phosphorylation.
NB cell line is used for this study. NB is the most common extracranial solid tumor of childhood, accounting for 15% of childhood cancer deaths. The non-differentiated stem cell phenotype may potentially be an independent risk factor for NB malignancy. Although incremental improvements in outcomes have been achieved with conventional chemotherapy agents, particularly RA, only one-third of children with NB are expected to survive long-term with the current chemotherapeutic interventions. The use of RA in NB has become the standard therapy.
As RA is tolerated fairly well as a single-agent, it may also function in combination therapy with other reagents. A clinical trial has been conducted using a combination of RA and SAHA that is the histone deacetylase inhibitor [34]. Another combination was explored with a combination of RA and ZD6474, a dual small molecule inhibitor of receptors for vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF) [35]. Despite its powerful chemotherapeutic effect, RA has been cautioned by producing toxic effects including chelitis and headaches. It has been previously reported that the patients receiving combination of RA and bexarotene, a chemotherapeutic drug for cutaneous T-cell lymphoma, produced fewer RA-associated toxic effects [36]. Combinations of retinoids with cytotoxic agents have shown enhanced antitumor efficacy in some animal models, including a combination of 9-cis-RA with cisplatin [37]. Therefore, a combination of RA therapy with other agents would be beneficial to reduce toxicity and increase antitumor efficacy. In this study, we demonstrated that the combination of RA and knock-down of DLK1, one of stem cell gene, enhanced NB cell differentiation (Fig. 2). This result provided another possible potential chemotherapeutic strategy for this deadly child cancer, NB. Since the proximal 5' promoter/ enhancer region of the DLK1 gene contains retinoic acid receptor elements, it would be interesting to characterize the mechanisms by which RA affects DLK1 transcription via the RA-dependent mechanism.
Elevated DLK1 expression is also reported in several tumor types, particularly neuronal tumors like NB and glioma [38]. Nonetheless, the role of DLK1 and its mechanism in tumor growth remains to be clearly elucidated. If DLK1 maintains tumor cells in a stem cell-like state, the relatively long lifespan of stem cells will allow undifferentiated tumor cells to accumulate and perpetuate stable genetic and epigenetic changes that ultimately result in tumor malignancy. The role of DLK1 in tumor progression needs to be investigated in further detail. In addition, there is a lack of evidence in the literatures demonstrating any other chemopreventive agents or dietary components that can down-regulate DLK1 expression. Currently, further researches on chemopreventive agents or dietary components that can affect DLK1 expression are underway in our laboratory.
Very little is known regarding the molecular mechanism and signal transduction pathway of DLK1 in cell differentiation. In a previously study, DLK1 was proposed to exert a potential impact on several membrane-associated signaling pathways that facilitate the maintenance or self-renewal of neuronal stem/progenitor cells such as EGFR, FGFR, and Notch, in addition to ERK phosphorylation [26]. To further provide the mechanism of signaling pathway of RA and DLK1 on NB cell differentiation, MAPK inhibitors, U0126 and PD98059, were used. In the present study, the phosphorylation of ERK during neuronal differentiation was shown to be induced by RA (Fig. 3) and MAPK inhibitors blocked neuronal differentiation induced by RA and DLK1 knockdown (Fig. 4 and 5). U0126 and PD98059 have been identified as specific inhibitors for the ERK/MEK1/2 pathway, and thus have been employed to study the involvement of the MAPK pathway in cellular events including differentiation [29,31]. Both U0126 and PD98059 are MEK inhibitors, and both are useful in probing the functions of MEK in a variety of biological processes, particularly those involving mitogen-activated protein kinase (MAP kinase/ERK) activation [39].
The findings of this study demonstrated that the differential effects of U0126 and PD98059 on neuronal cell differentiation induced by either RA or DLK1 knockdown (Fig. 4 and 5). The precise action mechanisms of U0126 and PD98059 as MEK inhibitors, and in particular as ERK inhibitors, remain a matter of some controversy. It has been reported previously that U0126 directly inhibits MEK1/2 activity, whereas PD 98059 does not directly inhibit MEK1 activity but does inhibit its activation (phosphorylation) by Raf-1 [29]. Furthermore, previous studies have demonstrated that U0126 inhibits MEK1 and MEK2, and PD98059 [29] inhibits MEK1, but only inefficiently inhibits MEK2 [40]. However, equilibrium binding studies showed that U0126 and PD98059 bind to MEK1 at identical or overlapping sites [40]. Dang and Lowik [41] characterized the differential effects of PD98059 and U0126 on osteogenesis and adipogenesis. However, this present study is the first to demonstrate that specific inhibitors of the ERK (p42/44 MAPK) pathway, PD98059 and U0126, exerted differential effects on the ERK phosphorylation induced by RA in NB. Therefore, even though there is clearly some overlap in the activities of U0126 and PD98059, U0126 evidenced more efficient inhibition than was observed with PD98059. This indicates that the neuronal differentiation induced by either RA or DLK1 knockdown requires the inhibition of both MEK1 and MEK2, as opposed to only one of each.
In conclusion, combination therapy is particularly important in cancer chemotherapy in regard to safety considerations. The findings of this study demonstrated that RA and DLK1 could provide a novel therapeutic as well as preventive strategy for regulating the differentiation of human malignant NB cells.





 XML Download
XML Download