

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Lung cancer is one of the most frequent types of cancer. There are 1.6 million new cases of lung cancer diagnosed each year. Based on cellular morphology, there are two main types of lung cancer: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). According to the National Cancer Institute (NCI), there will be 226,160 estimated new cases and 160,340 deaths from lung cancer (NSCLC and SCLC combined) in the United States, alone, in 2012.
The present approaches to lung cancer treatment include: surgery, chemotherapy, radiation therapy, targeted therapy, and immunotherapy. Until recently, immunotherapy of lung cancer, although considered safe, provided relatively little direct clinical benefit. Presently, no approved immune therapy exists for lung cancer. However, in recent years, substantial progress in our understanding of the mechanisms regulating immune responses in lung cancer has been made and new methods of immune therapy have been developed. A number of clinical trials have shown promise and, in recent years, a number of studies have demonstrated that a combined modality of cancer treatment can improve the outcome of standard treatment. This raises hope that substantial progress in the clinical application of immune therapy for lung cancer can be achieved in the near future.
The immune system uses various mechanisms against growing tumors, including cytotoxic T cells (CTL), natural killer (NK) cells, natural killer T cells, and, in some cases, antibodies. Dendritic cells (DC) play a central role in the antitumor immunity in solid tumors by coordinating the activities of the above mentioned cells. CTL employ several mechanisms to kill tumor cells. These include interferon-γ (IFN-γ), Fas/FasL (1), and perforin/granzyme B (GrzB) (2).
IFN-γ can induce programmed cell death and, together with interleukin (IL)-12 it was implicated in apoptosis via production of reactive oxygen species and nitrogen intermediates (3). IFN-γ can modulate a p53-independent apoptotic pathway by inducing select apoptosis-related genes like bak (4). IFN-γ also increased the sensitivity of tumor cell lines to tumor necrosis factor-α (TNF-α)-mediated and anti-Fas antibody-mediated cell death (4). It was reported that the combination of TNF-α/IFN-γ induces apoptotic cell death through a p53-independent, but c-myc-dependent pathway in a NSCLC cell line (5). The Fas/FasL complex involves FasL on the effector cells that engages the Fas receptor on the target cell. Upon activation by trimerization, Fas recruits adaptor molecules as Fas-associating protein with death domain, which activates caspase 8. This, in turn, activates caspase 3 that leads to DNA fragmentation (6,7). The Fas/FasL mediated apoptosis represents the extrinsic pathway that results in the activation of caspase 8. This caspase, either in response to FasL or TNF can induce the cleavage of Bid to yield a truncated carboxyl-terminal fragment that translocates from the cytosol to the outer mitochondrial membrane. Oligomers of the truncated Bid can trigger mitochondrial membrane permeabilization and cytochrome c-mediated caspase activation. This enhances intracellular signals that trigger DNA fragmentation via caspase-activated DNase (CAD). CAD release can also be initiated by non-caspase proteases, like cathepsins, calpains and granzymes (7). A more recent report provides the evidence on the importance of Kelch-like ECH-associated protein 1 (Keap1), a cytosolic inhibitor of NF-E2-related factor 2 (Nrf2), in targeting Bcl-2 degradation for apoptosis (8). Keap1, an adaptor protein that mediates the degradation of Nrf2, is a major regulator of expression of cytoprotective genes. Human lung cancer cell lines, with mutant Keap1, had decreased apoptosis in response to etoposide or UV radiation (8). Another mechanism for tumor cell killing involves the pore-forming protein, perforin, and granzymes, a family of serine proteases (9,10). Perforin induces formation of transmembrane channels. This leads to GrzB accessing intracellular substrates such as caspases. However, GrzB entry can also be mediated through receptor-dependent endocytosis. Perforin-expressing CTL play a major role in immune surveillance in hematologic malignancies (10). GrzB and perforin were found to be involved in the suppression of nodal metastasis in lung cancer. Both GrzB and perforin were distributed in the cytoplasm of cancer cells rather than in tumor-infiltrating lymphocytes even in early-stage tumors, and their expression was inversely correlated with the status of regional node metastasis (11). However, a more recent study on 67 patients with NSCLC and 30 with SCLC demonstrated that the GrzB and perforin A expression was actually reduced. NK cells in these patients had a decreased IFN-γ production and increased the immunoglobulin-like receptor (KIR) expression. This would potentially reduce cytotoxic function of these cells, in the presence of appropriate class I human leucocyte antigen (HLA) expression by tumor cells by delivering inhibitory signals to a greater proportion of NK cells. However, as tumor cell class I HLA is significantly down-regulated in cancer, increased inhibitory KIR expression becomes ineffective at blocking NK cell killing (12). Decreased IFN-γ was found not only in the NK cells, but also in the T cells from lung cancer patients (13,14).
MECHANISMS OF TUMOR ESCAPE FROM IMMUNE CONTROL
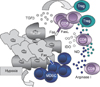
Tumors have a variety of mechanisms by which they evade host immune surveillance. These mechanisms affect a large variety of factors associated with tumor cells, tumor stroma, T cells, and antigen-presenting cells (15). These mechanisms include the lack of MHC class II and co-stimulatory molecules, down-regulation of MHC class I molecules and antigen processing machinery, secretion of immune suppressive factors like transforming growth factor-β, indoleamine 2,3-dioxygenase (IDO), etc. (Fig. 1) (16). These changes participate in the malignant cell evasion from T cell recognition and destruction. Various types of defects of the MHC class I antigens include: loss or down-regulation of HLA class I antigens and selective loss or down-regulation of HLA class I molecules (17). HLA class I defects were found with high frequency (93.6%) in NSCLC. Patients with NSCLC were segregated based on the expression of HLA class I molecules in primary lesions. Median survival in patients with negative staining for HLA class I was 40.6 months, with heterogeneous staining - 44.0 months, and with positive staining - 17.9 months, respectively. This raises a possibility that NK cells may play a prominent role in immune surveillance. Also, the frequency of HLA class I antigen loss was higher in adenocarcinomas (46.8%) than in squamous cell carcinomas (26%) (18). In contrast, a similar study from 161 patients with NSCLC found the association of the down-regulation of HLA class I expression with a poor prognosis. In this case the lesions' evaluation was performed by the use of a novel monoclonal anti-pan HLA class I heavy chain antibody (EMR8-5) allowing detection of HLA-A, -B, and -C antigens. Interestingly, unlike the previous study where no significant associations were found with any other clinical parameters, here it was reported that down-regulation of HLA class I significantly correlated with the male sex, a history of smoking, non-adenocarcinoma histology, and a moderate- or low-grade differentiation (19). Both of these studies used primary tumor specimens fixed in formalin and embedded in paraffin. The first study, however, analyzed patients with various stages of the disease (I~IV), and did not find an association between HLA class I expression and the stage of the disease; while the second study analyzed early-stage patients. This may account for different results. The other possibility could be the differences in racial make up of the study populations from the US (18) and Japan (19), since in the US there is a great heterogeneity in population in most of the clinical trials (20).
Tumors, including lung carcinoma, use FasL as a mechanism of immune evasion. Activation-induced cell death of memory CD8+ T cells (but not CD4+ T cells) from pleural effusion of lung cancer patients is mediated by the Fas-induced apoptotic pathway. Expression of FasL and tumor necrosis factor-related apoptosis-inducing ligand were up-regulated on CD8+ T cells from pleural effusion (Fig. 1) (21). As a result, there is a continuous renewal of lymphocytes at the tumor site without significant destruction of the tumor (22). However, in a mouse Lewis lung cancer model it was demonstrated that the lack of Fas on tumor cells can result in the promotion of tumor growth by the lack of FasL-mediated killing. These data shed new light on the role of Fas receptor in tumor progression. Fas can be upregulated by IFN-γ and/or TNF-α in many cell types (23). In another study, it was demonstrated that lung tumor growth in mice was promoted by Fas signaling through recruiting myeloid-derived suppressor cells (MDSC), via cancer cell-derived prostaglandin 2, thus linking the inflammation and immune escape mechanisms (24).
Inhibition of T cell function may also involve the down-regulation of CD3 T cell receptor expression on T cells (25). Lung tumors can down-regulate CD3ε expression on T cells without the induction of T cell apoptosis, via the Fas/FasL pathway. This effect was induced by soluble factors and was consistent with the data obtained on patients with lung adenocarcinoma. In patients, T cell dysfunction was produced by significant down-regulation of CD3ε expression on both, CD4+ and CD8+ T cells from pleural effusion, when compared to peripheral blood; and in the peripheral blood of patients as well, when compared to healthy donors (Fig. 1) (26).
In a recent study using NSCLC cell line Hasmin et al. (27), demonstrated that hypoxia-induced NANOG (a transcription factor associated with stem cell self renewal) promoted tumor cell resistance to antigen-specific lysis. NANOG expression is increased in tumor as compared with benign tissues. NANOG expression in NSCLC was associated with the up-regulation of cell replication and activation of STAT3 transcription factor.
Accumulation of regulatory T cells and MDSC, as well as immune suppressive macrophages and DC, contributes to the immune escape in cancer (Fig. 1) (28,29). Overall, tumor microenvironment blocks the ability of tumor-specific T lymphocytes to eliminate tumor cells; and, thus, not only limits tumor immune surveillance but also blunts the potential effect of immunotherapy. In the absence of the tumor microenvironment, functional potent, mature DC can be generated from the cancer patients' peripheral blood progenitors, CD14+ adherent monocytes, and can be used in immunotherapy as autologous antigen carriers (30). However, the effect of ex vivo generated DC still depends on the adequate function of endogenous DC, due to the fact that generation of the effective immune responses require antigen transfer between DC (31,32); and in cancer patients, the DC function is impaired in the peripheral blood as well as in the tumor draining lymph nodes (29,33).
TUMOR ASSOCIATED ANTIGENS IN LUNG CANCER
Although lung cancer is typically not considered as immunogenic tumor, a number of tumor associated antigens (TAA) were described in lung cancer. In 79 patients with lung cancer melanoma-associated antigen (MAGE)-3 was expressed in 42% of patients, sarcoma antigen NY-SAR-35 (33%), cancer-testis antigen NY-ESO-1 (30%), MAGE-1 (27%), cancer testis (CT) antigen CT-7 (20%), MAGE-4 (19%), CT antigen LAGE-1 (16%), and MAGE-10 (14%) (Table 1). Thirteen CT antigens were tested by reverse transcriptase-polymerase chain reaction; and out of these 73% tissues samples expressed at least one CT antigen. Other tissues co-expressed from 2 to 9 CT antigens. This study showed the multitude of existing lung cancer antigens. However, 27% of patients had no expression of CT antigens in lung tissues (34). Expression of MAGE-A3/6 was found in 50% of patients with NSCLC (including squamous cell carcinoma and adenocarcinoma); whereas NY-ESO-1 was expressed in 18.2% of cases (35). MAGE-A3/6 was expressed more in squamous cell carcinoma, while New York esophageal squamous cell carcinoma 1 (NY-ESO-1) in both squamous cell and adenocarcinoma. CT were higher expressed in male than female, and in advanced stage NSCLC patients positive for NY-ESO-1 showed worse survival when compared with NY-ESO-1 negative patients. No correlation between DC and CTL infiltration was found. However, higher infiltration of DC was observed in the patients with no expression of CT antigens. Correlation of the expression of CT antigens with the stage of the disease, in an extensive study on 239 patients with NSCLC, was reported (36). The frequency of MAGE-A4 expression was higher in stages II~IV than in stage I; and MAGE-A3/4 were associated significantly more frequently with squamous cell carcinoma than adenocarcinoma. Such correlation was not found for the cancer-testis antigen NY-ESO-1 or Kita Kyushu lung cancer antigen 1 (KK-LC-1). Expression of KK-LC-1 was detected in a similar percentage in patients with adenocarcinoma and squamous cell carcinoma (32.1% vs. 36.5%), while the expression of NY-ESO-1 varied from 8.2% vs. 15.9% in the same NSCLC clinical subtypes (36). MAGE-A4 positivity was found to be significantly associated with a poor prognosis (including in analysis limited to patients at stage I). Another study also found that the presence of MAGE-A3/4 and NY-ESO-1 was associated with poor prognosis (Table 1) (37).
Altered p53 function is present in most of the lung cancers (approximately 50%) due to single-point mutations or abnormalities in degradation of wild-type p53. In SCLC, mutant forms of p53 are present in more than 90% of patients. Thus, p53 can be considered a valuable TAA that can serve for immune recognition, and can be targeted by anti-p53-based cancer immunotherapy (38).
Survivin is an antiapoptotic protein that belongs to the inhibitory antiapoptotic protein family, and it is overexpressed in various cancers being correlated with a more aggressive disease, poor survival, and drug resistance. Because this protein is short-lived with a half-life of about 30 minutes, it has the potential to be overexpressed in the cytoplasm of tumor cells, and, due to its rapid degradation, the end result is an increased expression of survivin derived epitopes on the tumor cell surface (39). Survivin targeting was proved efficient in overcoming erlotinib resistance in epidermal growth factor receptor (EGFR)-mutation positive NSCLC (40). In another recent study of 62 patients with NSCLC, survivin expression was detected in over 50% of specimens. In normal lung tissues survivin was expressed in 10% of the specimens, and survivin expression was positively correlated with the presence of Bcl-2, another inhibitor of apoptosis, suggesting a possible synergy in the development, progression and metastasis in NSCLC (41).
Mucin 1 cell surface associated (MUC1) or polymorphic epithelial mucin is another tumor antigen that is highly expressed in lung cancer. High levels of mucin are correlated with a poor prognosis, reduced apoptosis (42), high metastatic potential, and immunosuppression in adenocarcinoma patients (43). This suggests that MUC1 can be used as a valuable biomarker not just a target for immunotherapy in lung cancer (Table 1).
Novel therapeutic targets are being discovered for lung cancer. A novel TAA, cell division cycle 45-like (CDC45L), was reported to induce CTL reactive to tumor cells in vitro and in vivo This antigen was found to be overexpressed in most of the lung cancer tissues from patients, but not in the non-cancerous ones, or in other normal tissues (44). In another study, ribosomal protein L19 (RPL19) was recognized by autologous CTL in lung adenocarcinoma. RLP19 was overexpressed in 40% of NSCLC tissues analyzed by immunohistochemistry, and was positively correlated with the production of IFN-γ by CTL. When suppressed with small interfering RNA, RPL19 induced tumor growth inhibition by suppression of cyclinD1, D3 synthesis, this effect being attributed to the cell cycle inhibition (Table 1) (45).
THERAPEUTIC APPROACHES TARGETING TAA IN LUNG CANCER
Depending on the nature of TAA, two major approaches are used for cancer immune therapy. The first approach utilizes defined TAA. In this case, the specific peptide representing TAA can be delivered to the patients either directly or in combination with different adjuvants, or loaded on DC. TAA can be delivered in the form of cDNA or RNA that are loaded, transfected, or transduced to DC.
Since MUC1 is expressed on the cell surface of many common adenocarcinomas, including lung cancer, it was used in immune therapy. A phase I study, using a modified vaccinia virus (Ankara) expressing human MUC1, which also contains a coding sequence for human IL-2 (TG4010), revealed a safe toxicity profile and some clinical activity (46). In 2 phase I multicentre studies, including 4 patients with lung cancer (3 with NSCLC and 1 with SCLC), patients were immunized with a modified vaccinia virus expressing human MUC1 (46). One of the lung cancer patients showed a marked decrease in the size of his metastases. This antigen-specific immunotherapy was tolerated by all 13 patients without major side effects. The cytoplasmic domain of MUC1 (MUC1-CD), that induces tumorigenesis and resistance to DNA-damaging agents, was found to be associated with poor outcomes in patients with lung adenocarcinoma (47). More recently, the therapeutic vaccination with TG4010 and first-line chemotherapy was tested in a controlled phase II trial in 148 patients with advanced stage NSCLC expressing MUC1. The results suggested that TG4010 could enhance the effect of chemotherapy, and a confirmatory trial has been initiated (48). Another novel approach of targeting MUC1 is the use of 86-base DNA aptamer (MA3) that binds to a peptide epitope of MUC1. In vitro experiments showed the specificity of MA3 for MUC1-positive tumors. When an aptamer-doxorubicin complex was designed this was found capable to carrying the drug to the MUC1-positive tumor cells. The drug intake, at the level of MUC1-negative cells, was significantly lower than in MUC1-positive cells (49). MUC1 has also been targeted in another trial of patients with NSCLC using the vaccine L-BLP25 (Stimuvax®) developed by EMD Serono Inc. and Merck. A multicentre study investigating the effect of vaccine in stage IIIB and IV NSCLC patients has initially demonstrated safety and a promising clinical effectiveness (50,51). This vaccine is being tested in a phase III randomized, placebo-controlled trial in stage III NSCLC patients (52).
Another protein vaccination strategy aims at MAGE-3. First results, reporting the successful induction of humoral and cellular immune responses in patients with NSCLC following vaccination with MAGE-3 with and without adjuvant chemotherapy, were published in 2004 (53). A recent review of NSCLC vaccines suggested MAGE-A3 as one of the promising alternatives for adjuvant therapy in lung cancer (54).
A number of therapeutic trials targeted antigens found to be expressed in lung cancer. In a phase I study reported in 2011, vaccination of patients with a NY-ESO-derived peptide that includes multiple epitopes recognized by antibodies, CD4 and CD8 T cells the increase in the titer of NY-ESO-1 antibody was detected in nine out of ten patients. CD8 and CD4 T cells responded with distinct specificity in all patients. Two patients with lung cancer showed stable disease (55).
Our group designed a p53 DC-based vaccine, using adenoviral vector (INGN-225) that was used in patients with extensive stage SCLC. Fifty four patients were treated in the initial trial (56,57). We found that the DC-p53 vaccine was safe, and able to induce significant immune response (in 57.1% of patients). Four objective clinical responses to the vaccine were registered. Most of the patients progressed. However, an unexpectedly high level of clinical responses was detected in patients after the treatment with salvage chemotherapy. This study helps to develop a novel paradigm of cancer treatment by the combination of immune therapy and chemotherapy. This paradigm will be discussed below. Based on the results of this study, we designed a randomized phase II trial in patients with SCLC, which is currently ongoing. It comprises 3 arms: patients treated with standard of care chemotherapy only, vaccine and chemotherapy, and a third arm where patients were treated as in the second arm plus all-trans retinoic acid (ATRA). The rationale of using ATRA is based on its effect on promoting the differentiation of MDSC, removing their immunosuppressive effect (58), and dramatically reducing the MDSC number, which improves the effect of tumor vaccines (59,60).
A second approach uses tumor cells as a source of TAA. In this case, it is expected that tumor cells provide large variety of not yet identified antigens. Those TAA can be delivered as tumor cell lysates, irradiated autologous, or allogeneic tumor cells. The second approach is illustrated by the use of GVAX vaccines. It is based on the fact that the growth factor granulocytes-macrophage colony-stimulating factor (GM-CSF) has been shown to induce antitumor immunity in a number of preclinical models (61). This prompted the strategy of using tumor cells, modified to secrete GM-CSF. It is expected that GM-CSF provides sufficient signal to attract DCs and stimulate the immune response, whereas tumor cells would be the source of TAA. Two early-phase clinical trials using GM-CSF-secreting autologous tumor cells (GVAX) in patients with NSCLC have shown encouraging preliminary results. Salgia et al. [62] reported on safety and feasibility of this approach in 33 advanced NSCLC patients with the most common toxicities being local injection site reactions and flu-like symptoms. A mixed response in one patient and long recurrence-free intervals in two other patients following isolated metastectomy were observed (62). In another phase I/II trial using the GVAX platform, autologous tumor cells were transduced with GM-CSF through an adenoviral vector (Ad-GM) and administered as a vaccine (63). Seventy eight percent of patients developed antibody reactivity against allogeneic NSCLC cell lines. Three durable complete responses were observed. More recently, in a phase I/II trial on advanced-stage NSCLC, autologous tumor was mixed with an allogeneic GVAX vaccine. Although objective tumor responses were not seen, the evidence of vaccine-induced immune activation was demonstrated with minimal toxicity (64).
A novel therapeutic approach, in patients with NSCLC, is the direct vaccination with messenger RNA (mRNA) encoding tumor antigens. Aimed to be used in combination therapies, this is a potent cancer vaccine that can facilitate the response to standard treatment. This vaccine can induce immune response, consisting in antigen specific CD4+ and CD8+ T cells and B cells. Clinical data were obtained from a phase I/II trial with promising results (65). These results may provide a good starting point for multimodality therapy in lung cancer.
REGULATION OF IMMUNE RESPONSES AND TUMOR MICROENVIRONMENT AS A WAY TO IMPROVE THE EFFECT OF CANCER IMMUNE THERAPY
Since tumor microenvironment is highly immune suppressive, it was logical to complement attempts to immune therapy of cancer with methods regulating the function of T cells. A number of these strategies currently reached clinical trials stage.
CTLA4 is a major negative regulator of T-cell function. It plays a role as a check-point limiting the uncontrolled expansion of T cells and thus preventing autoimmune abnormalities. The human monoclonal antibody against CTLA4 (Ipilimumab) was found to be effective in pre-clinical studies in combination with cancer vaccines (66). Treatment with ipilimumab increased antigen-specific CD8+ T cells in prevaccinated patients with melanoma (67). Ipilimumab demonstrated clinical efficacy in metastatic melanoma (67,68) and is currently approved for use in this disease. At this moment, no data are available regarding potential efficacy of ipilimumab in lung cancer. This issue deserves further investigation.
Programmed cell death protein 1 (PD-1) is another T-cell inhibitory receptor important for regulation of immune responses. PD-1 blockade was shown to enhance the effect of immune therapy in pre-clinical settings (69). In recent years a fully human IgG4 anti-PD-1 blocking antibody (MDX-1106) was used in a multicentre trial to evaluate safety, antitumor activity, pharmacokinetics, and immunological correlates in patients with refractory metastatic NSCLC, renal cell carcinoma, melanoma, or prostate cancer. The antibody was well tolerated with clear antitumor activity at 0.1 to 10 mg/kg dose when administered every 2 weeks for 8 weeks (in 12 cycles). Within this trial a maximum tolerated dose was not reached. The patients with NSCLC (squamous and nonsquamous) received doses of 1 to 10 mg/kg body weight (70). IDO is a tryptophan-catabolizing enzyme with immune-regulating activities in cancer progression. Regulation of IDO activity with 1-methyltryptophan demonstrated potent activity in preclinical experiments (71). Several clinical trials including those in lung cancer are underway to assess possible clinical utility of this approach.
MDSC is a major component of tumor microenvironment. These cells not only inhibit antitumor immune responses but also make tumor cells resistant to CTLs via production of peroxynitrite (72). Several clinical trials in different types of cancer, including lung cancer, are currently exploring the possibility of inhibiting MDSC and thus improving tumor-specific immune responses (29).
Chemotherapy, the conventional treatment option in patients with advanced stage lung cancer, dramatically affects tumor microenvironment. It debulks the tumor making it more accessible to T cells and should remove many of the immune suppressive factors mentioned above. However, chemotherapy has major immune suppressive effects rendering it rather detrimental to the immune system. Most of chemotherapeutic drugs target rapidly dividing tumor cells. Therefore, dividing cells of the immune system are also vulnerable to these drugs. Intensive chemotherapy causes lymphopenia with decreased percentage of circulating T cells (73). There is ample evidence that conventional chemotherapy can ablate T cell function and thus blunt anti-tumor immune responses (74). Therefore, for many years, the combination of immunotherapy and chemotherapy was not considered as very promising approach. In recent years, number of studies demonstrated clinical benefits for patients treated with cancer vaccines followed by chemotherapy (57,75-78).
Recently, we found that three chemotherapeutic drugs: taxol (TAX), doxorubicin (DOX) and cisplatin (CIS) sensitized tumor cells to the cytotoxic effect of CTLs (79), despite the fact that these drugs have very different mechanisms of action (80-82). This effect was associated with dramatically increased permeability of the target tumor cells to GrzB (79). Inhibition of GrzB activity abrogated the effect of TAX on CTL-induced apoptosis, confirming the critical role of this mechanism in the synergistic effect of chemotherapy and CTL (79). As we discussed above, perforin allows GrzB penetration into the cell (6,83). However, it has been also shown that penetration of GrzB into cells can be mediated by receptor-mediated endocytosis (84,85). One of the major receptors mediating GrzB uptake by cells is the multi-functional cation-independent mannose 6-phosphate receptor (CI-MPR) (86).
The two mannose-6-phosphate receptors, the 46 kDa MW cation-dependent (CD-MPR) and the ~300 kDa MW cation-independent (CI-MPR), are integral membrane proteins. On the cell surface CI-MPR, along with CD-MPR, is responsible for the binding and uptake of mannose-6-phosphate containing molecules (87). Within the cell, CI-MPR delivers the ligand-receptor complex from the trans-golgi network to endosomes, where the ligands are subsequently transferred to lysosomes (88,89). Some findings suggest that CI-MPR may play an important role in GrzB mediated cell killing (90,91).
We found that chemotherapy up-regulates the expression of CI-MPR on tumor cells and thus enable penetration of GrzB into tumor cells in perforin-independent manner (79). These data emphasize that chemotherapy may regulate GrzB uptake via up-regulation of CI-MPR and bypass the requirement for perforin. This led us to the conclusion that activated CTL, by releasing GrzB, were capable of killing the chemotherapy sensitized cells at the tumor site even in the absence of antigen expression. Based on these findings, we suggested a working hypothesis describing the synergistic effect of CTL and chemotherapy in cancer. Immunotherapy alone provides only limited number of tumor-specific CTL able to reach the tumor cells. Interaction of CTL with their target results in CTL activation, release of perforin and GrzB, and killing of tumor cells. However, only tumor cells that express antigen and are able to contact CTL will be killed. Thus, the effect of immunotherapy is limited by the number of CTLs able to penetrate tumor and by the number of tumor cells expressing specific antigen. This process is further compromised by different immune suppressive cells present at the tumor site. Chemotherapy causes the disruption of tumor stroma that allows more CTL to penetrate into tumor site. It also may inhibit negative regulatory network inside the tumor. Most importantly, chemotherapy up-regulates the expression of CI-MPR on tumor cells. As a result GrzB released by activated CTL can be picked up by a large number of neighboring tumor cells regardless whether they express tumor antigen or not. Thus, a relatively small number of CTL can cause apoptosis in large numbers of tumor cells manifested in a clinically evident antitumor effect. If correct, this hypothesis suggests that combination of chemotherapy and immunotherapy can be used as first line therapy in combined modality treatment schemes for advanced stage cancer. More studies, however, are needed to verify the validity of this hypothesis.
CONCLUSION
Cancer immune therapy went a long way from vague promise to clinical reality these days. We learned how to generate reasonably potent cancer vaccine and to prepare T cells for adoptive therapy. The challenge now is to develop strategies to overcome the negative effect of immune suppressive network present in cancer patients. Recent successes in the regulation of tumor microenvironment indicate that this challenge will be successfully met and cancer immune therapy will become an active part of therapy of lung cancer.

 XML Download
XML Download