

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Fluid and electrolyte disorders are among the most common clinical problems encountered in the setting of intensive care. Critical disorders such as severe burns, trauma, sepsis, brain damage, and heart failure lead to disturbances in fluid and electrolyte homeostasis. Possible mechanisms include reduced perfusion to the kidney due to hypovolemia or hypotension; activation of hormonal systems such as renin-angiotensin-aldosterone system and vasopressin; and tubular damage caused by ischemic or nephrotoxic kidney damage, including renal insult caused by a myriad of medications used in the intensive care. In addition, inappropriate administration of fluid and electrolytes should be considered in the diagnosis and treatment of fluid and electrolyte disturbances.
This article is intended to provide readers with relevant information on fluid and electrolyte problems frequently found in the intensive care unit (ICU), as well as on medications associated with fluid and electrolyte disorders.
Go to : 
Fluid management
Volume resuscitation of a patient with hypovolemic shock or sepsis is an essential component of patient care. Massive amounts of intravenous fluid are usually administered to replace intravascular volume deficit and to minimize complications attributed to hypovolemia such as tachycardia, hypotension, acute kidney injury, and multiorgan failure. Goal-directed therapies focused on restoration of normal blood pressure and organ perfusion have been advocated in the management of critically ill patients. Early goal-directed therapy, which is instituted in the initial phase of management of patients with severe sepsis or septic shock, has been shown to improve overall survival1). Clinicians should bear in mind that assessment of hemodynamic response to volume resuscitation and vasopressors should be based on specific hemodynamic and oxygenation parameters such as mean arterial pressure, central venous pressure, and central venous oxygen saturation, not solely on symptoms and physical findings.
In contrast to the notion of aggressive and liberal volume resuscitation, a growing body of evidence strongly suggests that fluid overload may be detrimental to critically ill patients. Relatively little attention has been paid to the consequences of fluid overload such as respiratory failure, increased cardiac demand, and peripheral edema. Recent studies on patients with acute lung or kidney injury have reported that fluid overload has been associated with adverse outcomes2-4). Although uniform definitions of fluid overload and well-designed randomized clinical trials are lacking, there seems to be a need to avoid overzealous fluid resuscitation in a subset of patients5).
As a general rule, daily input and output of fluid should be closely monitored, and loss into "third spaces" should be taken into account. Vital signs, findings from physical examination, and chest radiographs are of great importance in assessing the volume status of the patient. Invasive monitoring of central venous pressure or pulmonary capillary wedge pressure may be useful. Novel techniques involving invasive monitoring of extracellular fluid volume have been proposed, but none of them have been rigorously validated in clinical care6).
Go to :
Hyponatremia
Disturbances in plasma sodium concentrations are a common clinical problem in patients admitted to the intensive care unit. Many cases of dysnatremia are acquired after a patient is admitted to the ICU, and the presence of dysnatremia is associated with poor prognosis. A recent study involving 151,486 adult patients from 77 intensive care units over a period of 10 years has demonstrated that many cases of dysnatremia are acquired in the intensive care unit, and that the severity of dysnatremia is associated with poor outcome in a graded fashion7). Another study on the ICU patients with dysnatremias corroborated these findings, reporting that ICU-acquired hyponatremia and ICU-acquired hypernatremia were associated with increased mortality8).
Low plasma [Na+] represents a relative water excess in conjunction with impaired ability of the kidney to excrete electrolyte-free water. Removal of excess water by the kidney requires urinary dilution, which is compromised in virtually all patients in the ICU: (1) Heart failure, sepsis, shock, and multiple organ dysfunction syndrome impair glomerular filtration and enhance sodium and water reabsorption at the proximal tubule, thereby diminishing delivery of the filtrate to the diluting segment, i.e., the thick ascending limb of the loop of Henle and the distal convoluted tubule; (2) loop diuretics, thiazides, osmotic diuretics, and tubulointerstitial pathology reduce the reabsorption of sodium and chloride in the diluting segment; (3) and nonosmotic stimuli for vasopressin production such as pain, nausea, medications, and hypovolemia lead to increased water reabsorption in the collecting duct. In addition to impaired urinary dilution, hyponatremia in the critical care setting is related to inappropriate administration of hypotonic fluid9).
Symptoms of hyponatremia occur most commonly with a rapid decrease in plasma [Na+] to < 125 mEq/L. Seizure and coma usually result from rapid decrease in plasma [Na+] to < 110 mEq/L. The most dreaded complication in a patient with symptomatic hyponatremia is acute cerebral edema. Risk factors for development of acute cerebral edema should be identified before initiating therapy, including hypoxia, postoperative premenopausal women, elderly women on thiazide diuretics, polydipsic patients, children, and marathon runners10). Symptoms of hyponatremia may not be apparent in ventilated and sedated patients in the ICU, and worsening of cerebral edema may lead to catastrophic consequences such as brainstem herniation and respiratory arrest.
The time of development of hyponatremia and the presence of symptoms should dictate the management of hyponatremia. Correction of plasma [Na+] should be undertaken without delay in symptomatic patients, particularly those experiencing seizures. Development of hyponatremia in less than 48 hours and the presence of symptoms strongly suggest that the benefit of treating acute cerebral edema outweighs the risk of treatment-associated adverse effects. Hypertonic sodium chloride with or without a loop diuretic is usually started at a rate of 1-2 mL/kg/hr to raise sodium concentration by 1-2 mEq/L/hr. This rapid correction of hyponatremia should be limited to the initial phase of management. The overall correction of [Na+] should not exceed 8-12 mEq/L for 24 hours, as the risk of osmotic demyelination rises above this limit.
Estimated change in plasma [Na+] following the administration of 1 L of an intravenous fluid regimen can be calculated by equations proposed by Madias and Adrogue as follows:
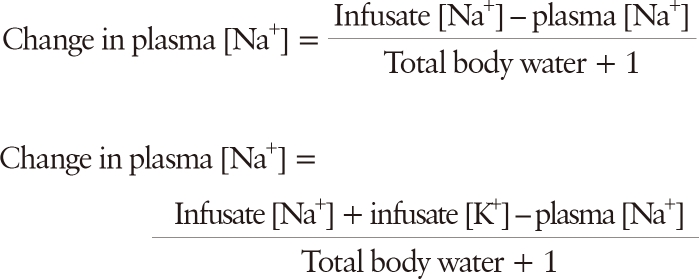
These equations have been shown to accurately predict changes in plasma [Na+] under most clinical settings with a tendency to underestimate the achieved plasma [Na+], sometimes by significant amounts11, 12). An important caveat to be remembered is that these equations assume human body as a closed system and therefore do not take into account ongoing fluid and electrolyte gain or loss. Thus, a cornerstone of therapy is close monitoring of symptoms, amount of fluid given to the patient, urine output, and plasma and urine electrolytes.
Euvolemic asymptomatic hyponatremia does not require urgent therapy, since the absence of symptoms indicates that brain cells have adapted to hypoosmolality. Identification and removal of reversible causes should be the first step of management.
Hypovolemic hyponatremia results from the loss of both water and solutes, with a greater relative loss of solutes. It is typically observed in a patient who was on a low-salt diet and was taking thiazide as an antihypertensive medication. Withdrawal of thiazide and replacement of volume deficit remove the stimulus for vasopressin release and initiate water diuresis. Plasma [Na+] should be closely monitored, since the onset of water diuresis may herald an abrupt rise in plasma [Na+]. In this regard, the initial choice of intravenous fluid should be 0.9% sodium chloride, unless the patient is symptomatic or hyponatremia is documented to have developed acutely (< 48 hours).
Hypervolemic hyponatremia occurs when both water and solute are retained in the body, but water is increased to a greater extent. Many cases of hypervolemic hyponatremia are associated with severe and sometimes irreversible dysfunction of the heart, liver, or kidney. Treatment of underlying disorders, restriction of both water and sodium intake, and administration of loop diuretics are helpful. Severe hyponatremia in patients with decompensated heart failure may require extracorporeal ultrafiltration, which has been consistently shown to improve congestion, lower diuretic requirements, and correct hyponatremias13).
To avoid osmotic demyelination during the treatment of hyponatremia, the rate of correction of plasma [Na+] should be less than 8-12 mEq/L/day. Patients who are at greater risk of developing osmotic demyelination include alcoholics, malnourished patients, hypokalemic patients, burn patients, patients with previous hypoxic episodes, elderly women on thiazide diuretics, and patients with plasma [Na+] < 105 mEq/L10).
Although V2 receptor antagonists are a useful option for management of euvolemic and hypervolemic hyponatremia, their use is associated with high cost of care, development of thirst which makes correction of hyponatremia more difficult, and the risk of rapid rise of plasma [Na+].
Go to :
Hypernatremia
Patients in the ICU are at a high risk of developing hypernatremia. Predisposing factors include the administration of sodium bicarbonate solutions to correct metabolic acidosis; renal water loss through a concentrating defect from renal disease or the use of diuretics or solute diuresis from glucose or urea; gastrointestinal fluid losses through nasogastric suction and lactulose administration, and water losses through fever, drainages, and open wounds. Acute diabetes insipidus with hypernatremia can complicate traumatic brain injury, occurring within 5-10 days after brain injury and disappearing within a few days to 1 month14).
Detection and treatment of hypernatremia require recognition of symptoms, identification of the underlying defects of water metabolism, correction of volume disturbances, and correction of hypertonicity. Primary manifestations are changes in mental status, including restlessness, irritability, lethargy, confusion, and somnolence. Patients may complain of intense thirst if they can communicate. Polyuric patients should undergo evaluation for defects in urinary concentration because previous knowledge of such disorders allows clinicians to adequately replace urinary water loss and avert the development of serious hypernatremia. Determination of urinary osmole excretion rate is helpful in differentiating water diuresis from solute diuresis. Patients at risk of developing diabetes insipidus include those with central nervous system disease and those receiving lithium or amphotericin B.
The rate of correction of hypernatremia depends on its rate of development and on the presence of symptoms. It is generally recommended that half of the water deficit be replaced in 12 to 24 hours as the neurological status is carefully monitored. The remaining deficit can be corrected during the next 48 hours. The maximum rate of plasma [Na+] decrease should not exceed 2 mEq/L/hr. Prescribed rate of correction can be lowered with improvement in symptoms. Ongoing loss of water during the course of treatment should be accounted for in the course of treatment. It is safe to measure plasma and urine electrolyte every one or two hours in the initial phase of management.
Hypervolemic hypernatremia is not uncommon and often iatrogenic, particularly in patients who receive large amount of saline or bicarbonate in the course of their illness. Treatment of hypervolemic hypernatremia requires removal of excess sodium from the body. Exogenous source of sodium should be eliminated unless clinically indicated. Loop diuretics with replacement of free water may be helpful in inducing negative sodium balance. Because hypervolemic hypernatremia is often an iatrogenic complication that develops only if renal function is compromised, renal replacement therapy is usually the only effective treatment in this setting. Composition of hemodialysis solution should be adjusted to lower sodium concentration. In terms of prevention, a rising plasma [Na+] should be regarded as a contraindication to further administration of saline, and should prompt treatment with hypotonic fluid via oral or parenteral route.
In treating patients with hypovolemic hypernatremia, correction of effective circulating volume deficit should be done before the replacement of water deficit, especially in patients with hypotension and obvious signs of hypovolemia. Isotonic sodium chloride should be given, and the underlying cause of volume loss should be identified and corrected. Treatment of euvolemic hypernatremia should be focused on repleting water deficit and decreasing ongoing loss of hypotonic fluids.
Go to :
Hypokalemia
Major causes of hypokalemia include low dietary potassium intake, shift into the intracellular compartment, extrarenal potassium loss, and renal potassium loss. Medications commonly prescribed in the ICU are associated with hypokalemia9). Sympathomimetics, insulin, methylxanthines, and dobutamine drive extracellular potassium into cells by stimulating Na+,K+-ATPase. Diuretics increase renal potassium loss by inhibiting sodium reabsorption in the loop of Henle and in the distal nephron. Amphotericin B is well known for its capability of disrupting the function of the collecting duct, causing nephrogenic diabetes insipidus, renal potassium loss, and distal renal tubular acidosis. Nonreabsorbable anions such as some penicillins and aminoglycosides can cause hypokalemia via obligatory potassium loss into urine.
Signs and symptoms of hypokalemia are largely neuromuscular, including paralysis, weakness, nausea, vomiting, constipation, respiratory muscle weakness, and rhabdomyolysis. The most dreaded complications related to hypokalemia are cardiac arrhythmias, especially in patients with hypertension, myocardial infarction/ischemia, or heart failure. Electrocardio-graphic changes seen in patients with hypokalemia include ST-segment depression, T-wave flattening, T-wave inversion, and the presence of U waves.
In the initial approach to the patient with hypokalemia, the major objectives are to rule out an emergency for which therapy must take precedence. The presence of respiratory muscle paralysis or changes in electrocardiogram should prompt a clinician to begin emergent therapy. If emergent settings are not present, the basis for hypokalemia should be sought by assessing the rate of urinary potassium excretion. If it is appropriately low (i.e., 24-hour urine K+ < 20 mEq/day or random urine K+/creatinine < 15 mEq/g or 1.5 mEq/mmol), transcellular shift or extrarenal K+ loss should be suspected. If urinary K+ excretion is high, transtubular potassium gradient (TTKG), acid-base status, and the presence or absence of hypertension are helpful in differential diagnosis of hypokalemia due to renal potassium loss. A TTKG larger than 4 suggests that there is an increase in K+ secretion into the cortical collecting duct, i.e., a high K+ concentration in the cortical collecting duct.
Except for emergent settings, oral administration of potassium is always preferred. Replacement of potassium via parenteral route should be reserved for patients with severe hypokalemia presenting with electrocardiographic abnormalities. Rapid infusion of potassium (i.e., > 10-20 mEq/hr) requires a central venous catheter, as infusion via a peripheral line causes phlebitis and vein injury. Amount of the initial dose should be in the range of 40-80 mEq. Total amount of daily K+ replacement should be less than 240-400 mEq/day. Parenteral K+ replacement should be given in the form of dextrose-free vehicles, as dextrose infusion induces insulin secretion and prevents rapid correction of extracellular K+ deficit. Hypocalcemia and hypomagnesemia should be identified and corrected if hypokalemia persists despite adequate supplementation.
Go to :
Hyperkalemia
Renal failure, adrenal insufficiency, insulin deficiency and resistance, and tissue damage from rhabdomyolysis, burns, or trauma are predisposing factors for hyperkalemia in critically ill patients. A lot of medications used in the ICU can also cause hyperkalemia, including beta-blockers, inhibitors of renin-angiotensin-aldosterone system, potassium-sparing diuretics, heparin and its derivatives, trimethoprim, and non-steroidal anti-inflammatory drugs.
Succinylcholine is a depolarizing muscle relaxant frequently used in the intensive care. It binds to the nitoconic acetylcholine receptors located in neuromuscular junctions and depolarizes muscle membrane by inducing Na+ and Ca2+ influx into and K+ efflux out of myocytes. Critical illness and anatomical or functional denervation of muscle result in (1) increased distribution of acetylcholine receptors throughout the muscle membrane beyond the neuromuscular junction, (2) altered subunit composition of acetylcholine receptors, and (3) the expression of a newly identified class of acetylcholine receptors which have longer duration of potassium efflux into the extracellular fluid15). As a result, the use of succinylcholine in critically ill patients is associated with higher potential for precipitating acute hyperkalemia. The risk of hyperkalemia is particularly higher in patients with upper or lower motor neuron defects, functional or chemical denervation (e.g., muscle relaxants, hypermagnesemia, clostridial toxins), direct muscle trauma, and severe infection or inflammation15).
In the absence of life-threatening conditions such as ventricular arrhythmia, initial diagnostic and therapeutic attempt should be focused on exclusion of pseudohyperkalemia, identification of the source of transcellular potassium shift, and discontinuation of medications potentially associated with hyperkalemia. The next diagnostic step is to assess urinary potassium excretion, which should be high in the face of high extracellular [K+] if renal function is not compromised (i.e., > 200 mEq/day or 140 µmol/min). If there is relatively low urinary K+ excretion, calculation of TTKG should follow to determine the cause of impaired renal K+ excretion. A low TTKG may warrant the administration of 9α-fludrocortisone to differentiate between aldosterone deficiency and resistance to aldosterone.
Treatment strategy of hyperkalemia depends on the presence of emergent conditions. If any electrocardiographic abnormality attributable to hyperkalemia is present, emergency management should take precedence over diagnostic evaluation. Intravenous calcium gluconate is the first step of management to antagonize the depolarizing effect of hyperkalemia. The next step should be to facilitate the shift of potassium into intracellular compartment. Insulin with 50% glucose is most effective as it is associated with most rapid and biggest drop in plasma [K+]. Intravenous or inhaled albuterol may be used. Sodium bicarbonate should be avoided in patients with extracellular fluid volume overload and is usually not the first choice in the treatment of hyperkalemia, as data on the usefulness of sodium bicarbonate are equivocal16).
After acute management, long-term therapy of hyperkalemia should include the removal of potassium from the body. Dialysis is an effective modality, especially for patients with renal failure. Sodium polystyrene sulfonate is slow to exert potassium-lowering effect, and can cause sodium retention and bowel necrosis. Sources for potassium excess such as muscle injury and tissue ischemia should be sought and treated appropriately.
Go to :
Hypophosphatemia
Hypophosphatemia has been associated with critical conditions such as Gram-negative sepsis and open heart surgery17-19). The prevalence of hypophosphatemia is high in the ICU, reported to be observed in about 28% in critically ill patients20). Hypophosphatemia (plasma phosphate concentration < 2.5 mg/dL or 0.81 mmol/L) may result from decreased intestinal phosphate absorption, increased renal phosphate losses, and a shift of phosphate to intracellular space. Clinical features of hypophosphatemia are widely varied, since numerous cellular mechanisms require phosphate (e.g., synthesis of adenosine triphosphate and 2,3-diphosphoglycerate). Hypophosphatemia is associated with leukocyte, erythrocyte, and platelet dysfunction; muscular weakness, confusion, ataxia, seizures and coma; respiratory failure; cardiac arrhythmias and cardiomyopathy.
Critically ill patients have underlying conditions that predispose them to developing hypophosphatemia, including malnutrition and inadequate body phosphorus stores, acute respiratory alkalosis, diabetic ketoacidosis, use of diuretics, alcoholism, and vomiting or gastric losses. Acute respiratory alkalosis can reduce plasma phosphate concentration as phosphate enters muscle. Infusion of glucose and the effect of hormones such as insulin, glucagon, and cortisol can decrease plasma phosphate concentrations by redistribution into intracellular space. Refeeding and regenerative processes may result in severe hypophosphatemia because of phosphate uptake in anabolic tissue. Decompensated diabetes mellitus with ketoacidosis is associated with excessive phosphate loss due to osmotic diuresis. Administration of insulin, fluids, and correction of ketoacidosis may reveal phosphate deficiency and cause a sharp decrease in plasma phosphate concentration due to intracellular shift.
Plasma phosphorus concentrations in critically ill patients should be maintained within normal range (2.5-4.5 mg/dL), given the potential adverse effects of hypophosphatemia. Treatment of hypophosphatemia depends on the magnitude of hypophosphatemia and the presence of symptoms. Asymptomatic mild-to-moderate hypophosphatemia (1-2.5 mg/dL) can be treated with oral phosphate supplementation if the gastrointestinal tract is intact. Symptomatic or severe hypophosphatemia (< 1.0 mg/dL) should be treated with intravenous phosphate. Plasma phosphate concentration < 0.5 mg/dL is associated with a phosphate deficit of more than 3 g; in the presence of symptoms, this deficit is more than 10 g21). The required dose of initial intravenous phosphate may vary from 2.5 to 19.8 mg/kg22). Typically, 2-5 mg/kg of inorganic phosphate dissolved in 0.45% saline is given over 6-12 hours and repeated as needed. Less than 50% of the initial dose should be given for patients who are not being treated with continuous renal replacement therapy (CRRT). Patients on CRRT should receive higher initial dosages, as the amount of phosphorus being removed by CRRT can be enormous. Further supplementation should be guided by clinical response to the initial doses and by plasma phosphate concentration.
Go to :
Hypocalcemia
Hypocalcemia is one of the most frequent electrolyte abnormalities encountered in the ICU. Low total concentrations of calcium have been reported to be affecting as many as 90% of critically ill patients, and the prevalence of hypocalcemia measured as ionized calcium is estimated to be about 15-20%23). Hypocalcemia is associated with increased mortality in patients cared in the ICU24).
Most common causes of hypocalcemia include trauma, acute and chronic renal failure, sepsis, hypoparathyroidism, hypomagnesemia, vitamin D deficiency, and complexing with citrate, albumin, or infused phosphate. Dilution of plasma induced by administration of massive amounts of intravenous fluid in a resuscitative effort is reported to be an important cause of hypocalcemia in trauma patients25). Decreased bone resorption, calcium chelation, calcitriol deficiency, decreased secretion or action of parathyroid hormone with or without hypomagnesemia, and increased urinary calcium excretion contribute to drug-induced hypocalcemia9). Spuriously low concentration of calcium can be detected following the administration of gadolinium-based contrast, as gadolinium interferes with calorimetric-based calcium assays, while ionized calcium concentration measurements remain unaffected26).
The symptoms and signs of severe acute hypocalcemia include tetany, papilledema, and seizures. Chronic hypocalcemia causes neuropsychiatric manifestations such as emotional instability, basal ganglia calcification, and extrapyramidal disorders; skin manifestations such as brittle and grooved nails, hair loss, dermatitis, and eczema; and cardiovascular manifestations such as prolonged QT interval, ventricular arrhythmias, and congestive heart failure.
Intravenous calcium should be given as calcium gluconate or calcium chloride to correct acute symptomatic moderate (total serum calcium concentration 7.5-8.0 mg/dL) or severe (total serum calcium concentration < 7.5 mg/dL or ionized calcium concentration < 0.9 mmol/L) hypocalcemia. Calcium gluconate should be preferred for routine calcium maintenance and supplementation. Use of calcium chloride should be restricted to urgent and emergent situations, since calcium chloride provides three times more elemental calcium than calcium gluconate.
Severe and symptomatic hypocalcemia requires urgent management. Initially, 1,000 mg of calcium chloride or 3 g of calcium gluconate may be given over 10 minutes to control symptoms. Continuous infusion of calcium may be required, with close monitoring of serum calcium levels at least every six hours during the infusion. The infusion rate should not exceed 0.8-1.5 mEq/min because of the potential risk for cardiac arrhythmias. Typically, as an intermittent bolus dosage, 1-2 g of calcium gluconate is mixed in 100 mL of 5% dextrose or 0.9% sodium chloride and infused over 30 to 60 minutes. The dose may be repeated every 6 hours as needed until serum calcium levels have been normalized.
Hypocalcemia is often accompanied by other electrolyte and acid-base disorders. Hypomagnesemia should be sought and corrected if hypocalcemia is not properly corrected by repeated calcium administration. Although the exact mechanism of calcium-magnesium interaction is unknown, it is hypothesized that magnesium deficiency may impair the release or activity of parathyroid hormone. When metabolic acidosis is present, hypocalcemia should be corrected before acidosis because the treatment of acidosis decreases the concentration of ionized calcium, thereby precipitating problems such as tetany or cardiac arrest. Bicarbonate solution and calcium salt should be administered in different intravenous lines to avoid precipitation of calcium carbonate.
Chronic or asymptomatic hypocalcemia does not require intravenous calcium therapy. Concomitant hyperphosphatemia, especially in patients with renal failure, requires phosphorus-lowering therapies. If hyperphosphatemia is not present, oral calcium supplements with or without vitamin D may be used.
Go to :
Hypomagnesemia
Hypomagnesemia is frequently observed in critically ill patients, and its prevalence in the ICU is reported to be as high as 50%27). Severe hypomagnesemia can result in electrocardiographic changes, arrhythmias including torsades de pointes, seizures, coma, and death. Hypomagnesemia is associated with concomitant electrolyte disturbances such as hypokalemia and hypocalcemia.
Causes of hypomagnesemia include excess gastrointestinal or renal losses, surgery, trauma, infection or sepsis, burns, transfusion of blood products preserved with citrate, alcoholism, starvation or malnutrition, and certain medications (e.g. diuretics, aminoglycosides, amphotericin B, cisplatin, digoxin, and cyclosporine). Fractional excretion of magnesium is a helpful index for differential diagnosis of hypomagnesemia22).
Serum magnesium concentration in critically ill patients should be maintained at 1.5 mg/dL or higher. Patients with acute myocardial infarction may require higher concentrations (i.e., > 1.7 mg/dL) to prevent fatal cardiac arrhythmias. The goals of therapy should be to avoid or resolve symptoms, return the serum magnesium concentration to 1.5-2.4 mg/dL, and avoid hypermagnesemia. Serum magnesium levels do not reflect total body magnesium store, since only 1% of magnesium stores are found in the extracellular space. Although a study has reported that magnesium deficiency reflects a total body magnesium deficiency of 1.0-2.0 mEq/kg, treatment of hypomagnesemia is largely empirical and plasma [Mg2+] should be carefully monitored.
Intravenous administration is preferred in critically ill patients with severe (plasma [Mg2+] < 1 mg/dL) or symptomatic hypomagnesemia. Infusion time is critical, because magnesium distributes into tissues slowly and is rapidly eliminated by renal excretion, with up to 50% of infused magnesium excreted in the urine. Severe hypomagnesemia requires treatment with 32-64 mEq (up to 1.5 mEq/kg) of magnesium. Doses less than 6 g of magnesium sulfate are infused over 8-12 hours, and higher doses are given over 24 hours. Reduced dose should be prescribed for patients with renal impairment to prevent hypermagnesemia. Mild hypomagnesemia can be treated with oral replacement.
Go to :
Conclusion
Fluid and electrolyte abnormalities in critically ill patients can lead to fatal consequences. More caution to electrolyte disturbances should be exercised in intensive care because it is often impossible to adequately assess symptoms and signs of critically ill patients. To provide optimal management, clinicians should be knowledgeable about fluid and electrolyte homeostasis and the underlying pathophysiology of the respective disorders. In addition, intensivists should pay attention to the administered fluid and medications potentially associated with fluid and electrolyte disturbances.
Go to :
 XML Download
XML Download