

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The acid-base regulation is chiefly dependent on the control of net acid excretion by the kidney and CO2 excretion by the lungs. Renal acid-base homeostasis consists of two major processes, the reabsorption of filtered bicarbonate and the excretion of the hydrogen ion. The kidney excretes hydrogen ion through the processes of titratable acid excretion and urinary ammonium excretion1, 2). Quantitatively, urinary ammonium excretion is the primary mechanism of net acid excretion both under basal conditions and in response to acid loads1-3). The Understanding renal ammonium production and transport is of fundamental importance for understanding acid-base homeostasis.
The term "total ammonium" is used to denote the sum of NH3 and NH4+. In this review, because the vast majority of the total ammonia is in the form of NH4+ at the physiologic pH, we generally refer to total ammonia transport as "ammonium transport" and to total ammonia excretion as "ammonium excretion"4).
Go to : 
Renal ammonium production
The proximal tubule is the chief site of renal ammonium production. Glutamine catabolism in the proximal tubule generates NH4+ and also bicarbonate after complete catabolism of α-ketoglutarate to CO2 and H2O. Studies in microdissected tubules have demonstrated that proximal tubules from the acidotic rats produced substantially more ammonium than did tubules from controls5). These results illustrate that the production of ammonium is regulated according to the acid-base state. In addition, hormonal factors contribute to the stimulation by metabolic acidosis of ammoniagenesis in the proximal tubule. The circulating levels of glucocorticoids increase during metabolic acidosis6).
Go to :
Renal ammonium transport
Renal ammonium metabolism and transport involves integrated responses of multiple portions of the kidney, including specific transport mechanisms in the proximal tubule, thick ascending limb of the loop of Henle, and the collecting duct.
1. Ammonium transport in the proximal tubule
The proximal tubule secretes NH4+ into the luminal fluid primarily through the action of the apical sodium/hydrogen ion exchanger, NHE37, 8), and to a lesser degree through an apical barium sensitive potassium ion channel8-10). The activity and abundance of the apical Na+/H+ exchanger NHE3 is increased in the proximal brush border membrane during metabolic acidosis, which could be expected to contribute to the enhanced NH4+ secretion by the proximal tubule during this condition11). It was recently shown that stimulation of ammonium secretion by the proximal tubule during metabolic acidosis largely depends on Na+/H+(NH4+) exchange activity and on angiotensin II12).
2. Ammonium transport in thick ascending limb of Henle's loop
After ammonium is produced and secreted by the proximal tubule, it is then delivered into the renal medulla via the loop of Henle. These potential losses of luminal NH3 are minimized because more than 75 percent of the tubular fluid NH4+ is recycled within the medulla, thereby maintaining a high interstitial NH3 concentration3, 13, 14). The primary step in this process is reabsorption in the thick ascending limb by substitution of NH4+ for K+ both on the Na+/K+/2Cl- carrier and, to a much lesser degree, through the K+ channels in the luminal membrane3, 15). The luminal NH3 permeability in the thick ascending limb was lower compared to other nephron segments16). The low NH3 permeability limits the backflux of NH3 into the tubule lumen and thereby contributes to the overall efficiency of the NH4+ absorptive process. Partial dissociation into NH3 and H+ then occurs in the less acidic tubular cell. As a result, the NH3 formed within the cell will diffuse out across the basolateral membrane into the medullary interstitium. The countercurrent multiplication of ammonium generates the maintenance of a high medullary interstitial NH3 concentration which promotes secretion into the medullary collecting tubule.
Ammonium reabsorption in the thick ascending limb of Henle's loop is reduced by hyperkalemia and is enhanced by chronic metabolic acidosis due to increased NH4+ production in and delivery out of the proximal tubule14, 17). In the thick ascending limb of the loop of Henle, metabolic acidosis increases ammonium reabsorption through mechanisms that appear to be involved in increasing NKCC2 expression17, 18). In vitro incubation of rat medullary thick ascending limb fragments in suspension in an acid medium strongly enhanced the BSC1/NKCC2 mRNA and protein abundance and cotransport activity18). In addition, administration of the glucocorticoid dexamethasone to adrenalectomized rats stimulated BSC1/NKCC2 expression at the mRNA and protein levels19). The acidity of the surrounding environment and glucocorticoids may account for the stimulating effect of chronic metabolic acidosis on BSC1/NKCC2 expression.
3. Ammonium secretion in the collecting duct
The majority of urinary ammonium is secreted into luminal fluid in the region of the nephron distal to the micropuncturable late distal tubule20). This is a heterogeneous region, and includes portions of the distal convoluted tubule (DCT), connecting segment (CNT), initial collecting tubule, cortical collecting duct (CCD), outer medullary collecting duct (OMCD) and inner medullary collecting duct (IMCD). Accordingly, understanding the mechanisms of transepithelial ammonium transport across the cells that comprise these portions of the kidney is important.
The mechanism of ammonuim secretion in OMCD and IMCD is not completely clear at present. Since the first description of the concept, the process of transepithelial transport of ammonium in the collecting duct is thought to occur primarily through passive non-ionic NH3 diffusion21, 22). The fluid entering the collecting tubules has a relatively low NH3 concentration, because of its removal in the loop of Henle. The net effect is that there is a relatively large gradient favoring the free diffusion of interstitial NH3 into the tubular lumen, where it forms NH4+1, 13). The cell membranes in the collecting tubules are highly permeable to NH3 but have only a negligible permeability to NH4+23). As a result, interstitial NH3 can passively diffuse into the tubular lumen where it is then trapped as NH4+. The net effect is that NH3 is secreted into the lumen throughout the collecting tubules3).
Although in the past ammonium was believed to move across epithelia entirely by passive diffusion, an increasing number of studies demonstrate that specific proteins contribute to renal ammonium transport3, 24, 25). The basolateral Na+,K+(NH4+)-ATPase pump has been demonstrated to mediate NH4+ secretion in the rat IMCD isolated and perfused in vitro26). The secretory Na+/K+(NH4+)/2Cl- cotransporter BSC2/NKCC1 was recently reported to mediate K+- and NH4+-dependent chloride secretion, and thus to be involved in transepithelial solute transport27). BSC2/NKCC1 was shown to be up-regulated by chronic metabolic acidosis in collecting ducts of the rat28).
The most recent addition to our understanding of the molecular mechanisms of ammonium metabolism is the identification of the ammonia transporter family of proteins25, 29). Rh B glycoprotein (Rhbg) and Rh C glycoprotein (Rhcg) are expressed in the renal DCT, CNT and collecting duct, the sites where approximately 80% of urinary ammonium is secreted30-36). In conditions of increased single-nephron ammonium metabolism, such as metabolic acidosis and reduced renal mass, Rhcg expression increases, suggesting that Rhcg mediates and has an important role in renal ammonium transport32, 35, 36). However, the nature of the transported substrate (NH4+ or NH3) by Rh glycoprotein has been controversial37-39). It was recently shown that both global and collecting duct-specific Rhcg deletion altered renal urinary ammonium excretion, indicating that collecting duct ammonium secretion is, at least in part, medicated by Rhcg and not solely by lipid diffusion40, 41). In addition, in vitro microperfused collecting ducts of Rhcg-/- acid-loaded mice show reduced apical permeability to NH3 and impaired transepithelial NH3 transport, indicating a role of Rhcg as an ammonium transport protein mediating the net flux of NH3 in the collecting duct.
Both the rat and the human kidney express both apical and basolateral Rhcg immunolabel30, 32, 35, 36). Basolateral Rhcg expression increases in parallel with urinary ammonium excretion in both metabolic acidosis and reduced renal mass, suggesting that changes in basolateral Rhcg expression may contribute to renal ammonium metabolism35, 36). We recently demonstrate that the mouse kidney expresses both apical and basolateral Rhcg, which is similar to findings in both the rat and human kidney, although there are strain-dependent differences in the level of basolateral Rhcg expression in the mouse kidney42). These observations suggest that basolateral Rhcg may contribute to facilitated transcellular ammonium movement. Furthermore, basolateral Rhcg is likely to be the same protein as apical Rhcg, but is trafficked to the basolateral plasma membrane. Fig. 1 summarizes the major transporters involved in renal ammonium transport.
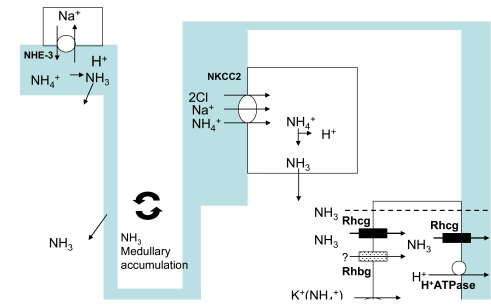 | Fig. 1Schematic representation of the ammonium transport mechanisms in the kidney. Ammonium is predominantly secreted into the luminal fluid via the apical Na+/H+ exchanger, NHE3. The thick ascending limb of the loop of Henle reabsorbs luminal ammonium, predominantly by transport of NH4+ by the apical Na+/K+/2Cl-cotransporter, BSC1/NKCC2. The renal countercurrent mechanism results in renal interstitial ammonium accumulation. Finally, the collecting duct secretes ammonium from the renal interstitium into the luminal fluid. NH3 is transported across the basolateral membrane, predominantly by Rhcg, but also by lipid diffusion (dashed line) and possibly by Rhbg. Intracellular NH3 is secreted across the apical membrane by apical Rhcg. In inner medullary collecting duct, basolateral Na+,K+-ATPase transports NH4+. There is also likely to be a component of diffusive apical NH3 transport (dashed line). H+-ATPase secretes H+, which combines with luminal NH3 to form NH4+. Modified from Kim et al.42).
|
In summary, renal ammonium handling involves intrarenal ammonium production and transepithelial transport in multiple tubular segments that results in highly regulated renal ammonium metabolism. An increasing number of studies demonstrate that specific proteins contribute to renal ammonium transport. Understanding the physiologic regulation of ammonium transporter and the contribution of other protein to renal ammonium transport, are likely to be important fields for future studies.
Go to :
 XML Download
XML Download