

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Electrolyte imbalance in neurologic patients
Electrolyte imbalance is known to be the most frequent systemic complication in patients with neurologic diseases1). Most electrolyte imbalances in neurologic patients, except dysnatremia, show comparable frequency with patients from other diseases and mainly influenced by medical conditions or co-morbidities2). For instance, hypokalemia is commonly manifested as a mild form combined with hypochloremic alkalosis and mostly associated with medical treatment such as diuretics, intravenous fluid, and iatrogenic hyperventilation. Meanwhile, hyperkalemia is almost always related to deterioration of renal function2).
Hyponatremia is the most common electrolyte imbalance in neurologic patients. It can be affected not only by the patient's own medical situation, but by neurologic disease itself though the disruption of arginine vasopressin (AVP) regulation3).
In this review, physiologic regulation of water and sodium balance, and characteristic sodium disturbances such as syndrome of inappropriate secretion of antidiuretic hormone (SIADH) and cerebral salt wasting (CSW) syndrome in neurologic patients will be described.
Physiologic regulation of water and sodium balance
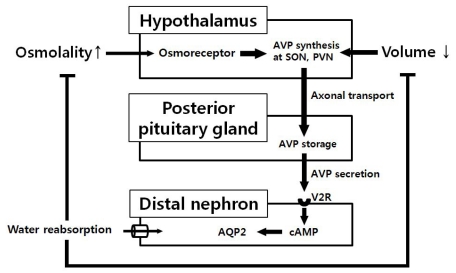
The kidney and the brain, with organ cross talk and interaction, play an important role in maintaining sodium and water balance4). AVP, also known as antidiuretic hormone (ADH), is a peptide hormone which is synthesized as a precursor prohormone in the supraoptic and paraventricular nuclei of the hypothalamic magnocellular neurons5). AVP is carried by its unique neurophysin from the hypothalamus to its storage site of axon terminals in the posterior pituitary gland. During the axonal transport, it is cleaved from the neurophysin and stored as a free form6).
The secretion of AVP is tightly regulated mainly by extracellular osmolality detected by osmoreceptors which are thought to be located in the anteromedial hypothalamus near the neurohypophyseal cell bodies in the supraoptic nucleus7). The secretion of AVP in response to plasma osmolality is extremely sensitive, and only 1% increase in osmolality above 280 mOsm/Kg can cause significant rise in plasma AVP levels, which can activate regulatory systems for antidiuresis and restore the plasma osmolality to normal8). At a plasma osmolality of 295 mOsm/kg, the plasma AVP level reaches approximately 5 pg/mL and maximum antidiuresis occurs, reflected by a urine osmolality greater than 800 mOsm/kg8). In addition to osmotic regulation, the secretion of AVP is also influenced by blood pressure and volume status detected by baroreceptors. Although the response of AVP to hypovolemia is less sensitive and does not occur without concomitant decrease in blood pressure, excess amount of AVP is secreted and acts as a potent vasoconstrictor on the vascular smooth muscle cells during the hypovolemic or vasodilatory shock9). Angiotensin II also have shown to induce AVP release in some animal studies via angiotensin II type 1 receptors in the lamina terminalis, located in the anterior wall of the third ventricle10). Nausea is another potent nonosmotic factor triggering the secretion of AVP to increase the plasma level by 20 to 500 fold even if the nausea is short-lived and unaccompanied by vomiting or changes in blood pressure7). After secretion, AVP circulates as a free form peptide, diffuses readily into the extracellular fluid space, and is metabolized within minutes by vasopressinase in the kidney and liver11).
AVP is an effective agonist to all vasopressin receptor isoforms: V1a, V1b, and V2 receptor (V2R). The antidiuretic effect of AVP is mediated by V2R expressed on the basolateral membrane of principal cells in the collecting tubule and epithelial cells of the thick ascending loop of Henle12). Binding of AVP to V2R in these cells activates adenylate cyclase, which in turn increases cytosolic cyclic adenosine monophosphate (cAMP) and cAMP mediated activation of protein kinase A (PKA). Subsequently, activation of PKA results in phosphorylation of aquaporin 2 (AQP2) and its translocation from intracellular vesicle to the apical membrane of the principal cells of the collecting tubule13). As a result, urine flow rate is reduced by increased transepithelial solute free-water reabsorption from the tubular lumen by this apical localized AQP2 which increases hydro-osmotic permeability of the tubular cells (Fig. 1).
Hyponatremia in patients with neurologic disorders
Hyponatremia defined as serum sodium level less than 135 mEq/L is known to occur in up to 20% of hospitalized patients14) and is associated with 1.5 fold higher in-hospital mortality rate than patients without hyponatremia15). Hyponatremia is more common in patients with neurologic disorders than in the general in-hospital population16) and the incidence is reported to be 34% of patients with aneurysmal subarachnoid hemorrhage (SAH)17) and more than 70% in tuberculous meningitis18). The prevalence of hyponatremia developed after transsphenoidal pituitary surgery for tumor resection may range between 25% and 35%19, 20).
In the presence of hypoosmolality by hyponatremia, water gain causes cerebral edema. Soon after this osmotic insult, the restoration of brain volume occurs through the adaptation mechanisms of brain cell, also known as 'osmoregulation', by losing the electrolytes and organic osmolytes21). However, patients with neurologic diseases may be more vulnerable to cerebral edema due to impaired osmoregulation in pathologic lesions of the brain22). Thus, these patients can exhibit more aggressive cerebral edema with deterioration of both clinical status and intracranial pressure than hyponatremic patients without brain lesions22). Furthermore, hyponatremia accompanied by central nervous system (CNS) disorders actually has shown to increase delayed cerebral ischemia and mortality rates23).
Although many etiologies such as diuretics, gastrointestinal salt loss, hypotonic solutions, and various comorbid conditions may cause hyponatremia, two main mechanisms of hyponatremia, SIADH and CSW syndrome, account for most of the hyponatremia in patients with neurologic diseases and is directly related to disruption of organ interaction between the brain and kidney24). Differential diagnosis between these two disease entities can be difficult due to considerable overlap in the laboratory findings and clinical situation.
SIADH in patients with neurologic diseases
SIADH is volume expanded status due to increased renal water reabsorption by excessive and inappropriate AVP secretion in the absence of hyperosmolality or volume depletion25). In addition, downward resetting of the osmotic threshold for thirst in these patients is known to be implicated in the development of volume expansion26). However, these patients do not show overt hypervolemic signs, because only one-third of the fluid remains in extracellular space. Nevertheless, increased glomerular filtration rate induced by this modest intravascular volume expansion lead to decreased proximal sodium reabsorption and increase urinary sodium excretion27). This sodium wasting accompanied with water retention often lead to an equilibrated state of sodium and water balance due to the vasopressin escape phenomenon28). Such normal renal sodium handling despite hyponatremia is a characteristic feature of SIADH. Serum levels of uric acid and urea nitrogen which co-transported with sodium in the proximal tubule are also decreased with increased sodium excretion, and return to normal value with correction of hyponatremia in patients with SIADH27, 28).
The diagnosis of SIADH is usually made from history, physical examination, and suitable laboratory tests. The diagnostic criteria29) for SIADH is summarized in Table 1.
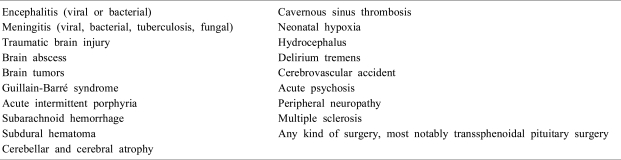
SIADH is associated with various neurologic diseases and neurosurgical procedures including encephalitis, meningitis, traumatic brain injury, brain tumors and trans sphenoidal pituitary surgery (Table 2)30). The stimulation of AVP release from the neurohypophysis in these diseases may be the possible mechanism for SIADH except some neuroendocrine tumors with ectopic AVP secretion31).
The treatment of hyponatremia in patients with SIADH should be guided by the severity of the disorder, onset and symptoms of hyponatremia32). Fluid restriction is the mainstay of therapy in patients with asymptomatic hyponatremia. Although patients with severe neurologic symptoms due to acute reductions in serum sodium, require rapid initial correction with hypertonic saline, excessively rapid correction should be avoided because it can lead to late onset of neurologic complications from osmotic demyelination29, 33).
The AVP receptor antagonist represents more targeted therapy to the treatment of hyponatremia in SIADH, and may be potentially beneficial for the treatment of hyponatremia in patients with SIADH secondary to neurologic diseases34). However, the exclusion of CSW syndrome through careful assessment of volume status and strict monitoring of serum sodium during treatment will be needed to avoid complications.
CSW syndrome in patients with neurologic diseases
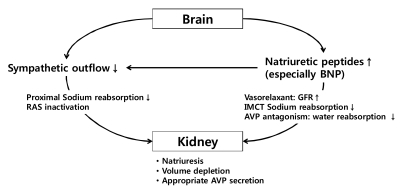
Many neurosurgical patients with hyponatremia, who meet the diagnostic criteria for SIADH, have a volume status incompatible with that diagnosis. The evidence of volume depletion and negative sodium balance estimated by mass balance for urinary electrolytes in these patients is more consistent with the diagnosis of CSW syndrome24). CSW syndrome, which is frequently followed by SAH, is characterized by excess renal sodium wasting with resulting volume depression35). Although, the mechanism of CSW syndrome is not fully understood, the most possible hypothesis is central amplification of natriuretic peptides, especially brain natriuretic peptide (BNP), combined with decreased sympathetic outflow due to various neurologic diseases36, 37) (Fig. 2). Since the sympathetic tone in the kidney plays an important role in proximal sodium and water handling and the control of renin release in juxtaglomerular epithelioid cells38), decreased sympathetic input into the kidney induces urinary sodium wasting and volume depletion. In addition, natriuretic peptides stimulate dilatation of afferent renal arterioles and constriction of efferent arterioles, leading to an increased glomerular filtration rate39). These peptides can also act on renal tubules, suppressing angiotensin II stimulated sodium and water transport, inhibiting sodium transporter in the inner medullary collecting tubules, antagonizing the renal effects of AVP and reducing sympathetic tone24, 40). Once volume depletion occurs due to renal sodium wasting, it activates baroreceptors that increase AVP release appropriately, resulting in increased water conservation. Therefore, most patients with CSW syndrome show elevated AVP levels and meet the criteria of SIADH. Nevertheless, distinguishing between CSW syndrome and SIADH should be emphasized since the adequate treatment regimen is quite different from each other23). Although, these disorders are commonly associated with neurologic diseases and there may be overlapping laboratory findings, volume status tends to be normal or slightly increased in SIADH, whereas decreased in CSW syndrome. Thus, the evidences of volume depletion such as weight loss, negative fluid balance, orthostatic hypotension and tachycardia in a hyponatremic patient with neurologic disease are strongly suggestive of CSW syndrome27). In addition, laboratory findings reflecting hemoconcentration such as elevated hematocrit, albumin, bicarbonate and urea nitrogen levels can provide further support for that diagnosis36). However, uric acid, which is usually elevated in patients with volume depletion, tends to be unexpectedly low in these patients41). As noted above, hypouricemia due to increased urate excretion also existed in patients with SIADH. But, the correction of serum sodium concentration usually leads to normalization of uric acid levels in patients with SIADH. On the other hand, hypouricemia persists after the correction of hyponatremia in patients with CSW syndrome, possibly due to prolonged proximal tubule dysfunction28, 42). Table 3 shows differential clinical features and diagnosis of CSW syndrome and SIADH.
In patients with CSW syndrome, the cornerstone of treatment is achieving euvolemia through vigorous volume resuscitation with intravenous saline and correcting hypotonicity with sodium replacement24, 27). Administration of mineralocorticoid agents which increases sodium reabsorption in renal tubules can also be considered in cases refractory to salt and fluid therapy43). As noted above, volume restriction following erroneous diagnosis of SIADH in patients with CSW syndrome can potentially aggravate hyponatremia. Thus, careful assessment of volume status in patients is needed to differentiate these diseases.
Summary
Hyponatremia is the most common electrolyte imbalance in patients with neurologic disease. SIADH and CSW syndrome are two main mechanisms of hyponatremia in these patients, excluding iatrogenic causes. Distinction between the diseases may be difficult due to overlapping laboratory findings and clinical presentations, but have critical importance since the adequate therapy is quite different each other and erroneous diagnosis can lead to grave prognosis as well as aggravation of hyponatremia. The fundamental differences between the diseases are the appropriateness of increased AVP release and presence of dysfunctional renal sodium handling. SIADH is in a volume expanded status due to inappropriately secreted AVP and water restriction is the treatment of choice. However, CSW syndrome is in a volume depleted status characterized by renal sodium wasting and appropriate secretion of AVP, requiring volume resuscitation combined with sodium replacement. Thus, physicians should focus on the volume status of hyponatremic patients with neurologic disease to differentiate between these two diseases.


 XML Download
XML Download