

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Renal dysfunction in chronic liver disease is characterized by impaired natriuresis, decreased free water clearance, and decreased glomerular filtration rate (GFR). Hyponatremia, ascites, and hepatorenal syndrome (HRS) represent the clinical consequences of disturbances in renal function. Optimal management of renal dysfunction in cirrhosis is extremely important in that renal dysfunction frequently complicates the clinical course of advanced liver disease and is invariably associated with poor clinical outcomes. Hyponatremia is present in about 50% of patients with cirrhosis and is associated with increased rate of other complications such as gastrointestinal bleeding, spontaneous bacterial peritonitis, and hepatic encephalopathy1). The presence of ascites predicts poor clinical outcome in cirrhotic patients, as shown by the 3-year survival rate for patients with ascites at 50%2). Progressive liver failure and superimposition of precipitating events culminate in the development of HRS, a state of severe intrarenal vasoconstriction and reduced GFR without intrinsic renal damage. Survival of patients with liver disease continues to be affected by the presence of renal dysfunction, even after they underwent liver transplantation3).
Renal dysfunction in cirrhosis is a clinical consequence of peripheral arterial vasodilatation and hyperdynamic circulation caused by portal hypertension. Clinical observations and recent experimental studies have shed light on the pathogenesis of hyperdynamic circulation in chronic liver disease. Better understanding of the pathophysiology enabled clinicians to introduce effective therapies for renal dysfunction once considered irreversible or medically intractable, and led to the proposal of new concepts and diagnostic criteria for HRS4).
Pathophysiology of renal dysfunction in chronic liver disease
1. Peripheral arterial vasodilatation and hyperdynamic circulation in chronic liver disease
Portal hypertension in cirrhosis is one of the best examples of hyperdynamic circulation, which results from a combination of increased cardiac output and dilated peripheral vascular bed5). Investigators have been aware of the importance of primary vasodilatation in cirrhosis for a long time, and bedside observation made by astute clinicians describes the typical features of hyperdynamic circulation in patients with cirrhosis, including increased pulse pressure, warm extremities, and capillary pulsations in the nail bed. Based on these findings, Kowalski and Abelmann were the first to demonstrate an increase in cardiac output and a decrease in peripheral vascular resistance in a patient with alcoholic cirrhosis6). A subsequent study corroborated this finding7). In addition, a host of studies using Doppler ultrasonography to assess blood flow in various organs in patients with cirrhosis have demonstrated (1) the state of hyperdynamic circulation in chronic liver disease, (2) predominant vasodilatation in the splanchnic vessels, and (3) relative vasoconstriction in other organs such as the kidneys8 ,9).
Splanchnic and systemic vasodilatation in the wake of portal hypertension creates a state of relative hypovolemia, which activates sodium-conserving mechanisms and leads to an increase in plasma volume. Most of the increase in plasma volume is used to fill up the increased splanchnic vascular compartment. Meanwhile, portal blood flow increases in the face of increased intrahepatic vascular resistance, as portosystemic collaterals partially decompress the portal vein and provides conduits for pooled splanchnic blood. The importance of increased blood flow in the portal vein was shown in a previous study, which reports that an increase in portal venous flow is a major contributing factor in maintaining and aggravating portal hypertension in conditions of increased intrahepatic resistance10).
2. Alterations in renal function
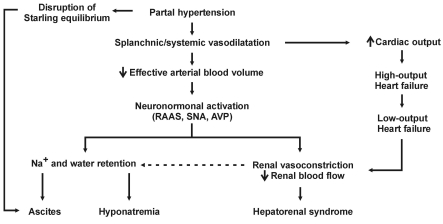
Splanchnic and systemic vasodilatation leads to renal vasoconstriction and impaired renal function (Fig. 1). Relative hypovolemia activates the sympathetic nervous system and renin-angiotensin-aldosterone system, and increases nonosmotic secretion of arginine vasopressin (AVP), resulting in sodium and water retention and development of ascites and hyponatremia. Renal hypoperfusion and cardiac dysfunction combine to provoke severe intrarenal vasoconstriction leading to HRS.
The clinical course of renal impairment in cirrhosis is further complicated by progressive cardiac dysfunction. As a result of peripheral vasodilatation and retention of sodium and water, cardiac output increases in the early phase of chronic liver disease. However, this increase in cardiac output fails to meet the need of the body and results in high-output cardiac failure. As peripheral vasodilatation continues to progress, myocardial contractility and cardiac output begin to decrease and low-output cardiac failure ensues11). Reduced cardiac output decreases renal perfusion and may precipitate the development of HRS, as renal hypoperfusion seems to trigger severe intrarenal vasoconstriction12).
3. Pathogenesis of splanchnic arterial vasodilatation
In vivo and in vitro studies have investigated the mechanisms of splanchnic vasodilatation in chronic liver disease by using experimental animal models for cirrhosis. Major findings from these studies are as follows: (1) the source of vascular hyporeactivity to vasoconstricting stimuli lies in the vascular endothelium13); (2) endothelial and neuronal isoforms of nitric oxide synthase (eNOS and nNOS, respectively) are upregulated in the splanchnic circulation14); (3) cirrhotic animals are remarkably sensitive to the effect of nonspecific inhibition of nitric oxide (NO) synthesis as compared with normal controls15); (4) and inhibition of NO synthesis almost completely normalize major hemodynamic abnormalities and renal function16,17). Vascular hyporesponsiveness and increased NO production were observed in major systemic arteries as well as in splanchnic arteries18,19).
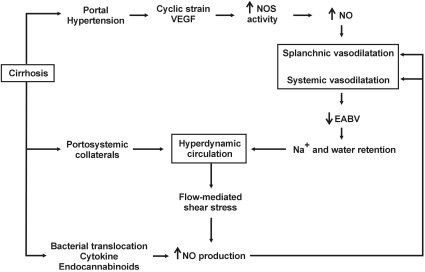
Nitric oxide is believed to be a key player in pathogenesis of splanchnic and systemic vasodilatation in chronic liver disease. Portal hypertension activates eNOS and nNOS in the splanchnic circulation through a myriad of putative mechanisms, including increased shear stress on the mesenteric arterial wall, increased expression of vascular endothelial growth factor (VEGF) in the splanchnic microcirculation, overproduction of inflammatory cytokines (e.g., tumor necrosis factor), and bacterial translocation (Fig. 2). Upregulation of NO synthases is not likely to be induced by increased mesenteric blood flow, as the time-course of NO production has shown that overproduction of NO occurs before the blood flow increases in the superior mesenteric artery20). Rather, other signals located upstream of NO, such as cyclic stress in the mesenteric arterial wall or increased expression of VEGF, seem to be involved in the enhanced eNOS activity. Additional studies lend support to the role of VEGF in splanchnic vasodilatation by showing that inhibition of VEGF receptor effectively attenuates splanchnic vasodilatation21,22).
Increased NO production leads to splanchnic and systemic arterial vasodilatation, which, combined with increased effective arterial blood volume, increases blood flow and augments NO production, as an increase in blood flow is a well-known stimulus for NO synthases (Fig. 2). As mentioned before, portosystemic shunts have a significant role in maintaining or aggravating hyperdynamic circulation, and increased portal blood flow can enhance the production of NO in the splanchnic circulation. Circulating hormones (e.g., endocannabinoids), gastrointestinal hormones (e.g., glucagon), and proinflammatory cytokines (e.g., tumor necrosis factor-alpha) induced by bacterial translocation can contribute to increased NO production23-25).
There are NO-independent mechanisms involved in hyperdynamic circulation in cirrhosis, as knockout of eNOS did not completely prevent the development of hyperdynamic circulation in portal hypertension26). Studies on impaired reactivity of the endothelium to vasoconstrictors in cirrhotic animals have reported reduction in phosphorylation of the myosin light chain (MLC) of the vascular smooth muscle cell (VSMC). Contractile agonists usually stimulate MLC phosphorylation via the activation of MLC kinase or the inhibition of MLC phosphatase. Phosphorylated MLC in turn activates actin-myosin ATPase, thereby crosslinking actin-myosin to induce smooth muscle contraction. Vasoconstrictors such as epinephrine and vasopressin bind to their respective receptors on the surface of the VSMC and activate phospholipase C (PLC), which produces inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 mobilizes calcium from the sarcoplasmic reticulum, and DAG activates protein kinase C which is involved in the increased activity of MLC kinase. In addition, receptor activation leads to increased activity of rho kinase, which is believed to inhibit the activity of MLC phosphatase. As a result, increased MLC phosphorylation and increased intracellular calcium concentration leads to smooth muscle contraction.
Clinical manifestations
Hyponatremia, ascites, and HRS are among the principal clinical consequences of the progressive arterial vasodilatation and hyperdynamic circulation in cirrhosis. Impaired sodium excretion and free water clearance result in the development of hyponatremia and ascites, and progressive decrease in GFR results in the development of HRS.
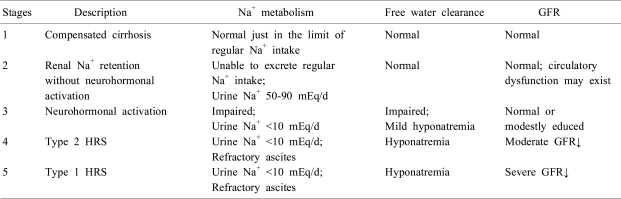
Progression of renal dysfunction in cirrhosis can be divided into five stages (Table 1)30). In the early phase of liver dysfunction (stage 1), patients can excrete daily sodium intake, though just in the range of normal daily sodium intake, and show normal GFR and absence of ascites. In the next phase of renal dysfunction (stage 2), impaired natriuresis takes place without overt activation of neurohormonal systems, as patients cannot mount an adequate natriuretic response to exogenous sodium load while concentrations of plasma catecholamine, aldosterone or AVP remain within the normal range. Increased expression of renal sodium transporters in the distal tubule seems to be involved in this phenomenon31). Subtle unidentified circulatory dysfunction also may play a role. As liver function deteriorates (stage 3), overt activation of neurohormonal systems occurs, and ascites and hyponatremia develop. In the final stages of cirrhosis, severe vasodilatation and renal vasoconstriction lead to refractory ascites and HRS. Type 2 HRS (stage 4) is considered to be a mild, slowly progressive form of renal failure, and type 1 HRS (stage 5) is characterized by rapidly worsening renal function, refractory ascites, and severe sodium retention.
Hyponatremia is associated in a graded fashion with other serious complications of liver cirrhosis. As compared with patients with mild hyponatremia (plasma sodium 131-135 mEq/L), patients with moderate-to-severe hyponatremia (plasma sodium ≤130 mEq/L) have significantly higher risk of developing hepatic encephalopathy, spontaneous bacterial peritonitis, and gastrointestinal bleeding1). The most common precipitating factor for hyponatremia is inappropriate use of diuretics, especially for patients with ascites but without peripheral edema. As the peritoneum has limited capacity for mobilizing ascitic fluid (~500 mL/day), excessive diuresis can reduce intravascular volume and renal free water clearance, leading to development of hyponatremia.
Ascites develops as a result of sodium retention by the kidney and disruption of the Starling equilibrium in the splanchnic circulation. As mentioned earlier, activation of the sympathetic nervous system, renin-angiotensin-aldosterone axis, and nonosmotic AVP secretion leads to sodium and water retention by the kidney. Increased hydrostatic pressure and decreased oncotic pressure in the capillary of the bowel, which occur as a result of portal hypertension, increases hepatic lymph production.
HRS is caused by severe renal vasoconstriction which occurs in patients with advanced liver disease and circulatory dysfunction. By definition, HRS is not associated with intrinsic renal disease or nephrotoxic injury, as kidneys from patients with HRS function perfectly when transplanted to other patients. HRS may occur spontaneously, but is frequently precipitated by bacterial infections such as spontaneous bacterial peritonitis (SBP), gastrointestinal bleeding, or inadequate albumin replacement after therapeutic paracentesis. These precipitating events abruptly reduce renal perfusion, thereby tipping the balance toward vasoconstriction between intrarenal vasodilators such as prostaglandins and vasoconstrictors. Imbalance between vasoactive mediators within the kidney further diminishes renal blood flow and causes HRS.
The diagnosis of HRS is based upon documentation of progressive renal failure and exclusion of intrinsic structural renal damage and other systemic illness affecting renal function. Type 1 HRS is a severe, rapidly progressive type of renal failure (i.e. doubling of serum creatinine to a level greater than 2.5 mg/dL in less than 2 weeks), while type 2 HRS represents moderate, slowly progressive renal failure (i.e. serum creatinine 1.5-2.5 mg/dL). Since the first proposal of diagnostic criteria in 199432), new concepts have emerged from experimental and clinical studies: (1) peripheral arterial vasodilatation predominantly occurs in the splanchnic vascular bed; (2) cardiac output in patients with HRS is insufficient for the patient's needs; (3) development of type 1 HRS is often triggered by other superimposing factors, most commonly spontaneous bacterial peritonitis or gastrointestinal bleeding; (4) renal function can be improved by medical treatment in patients with HRS4). As compared with the diagnostic criteria in 1994, new diagnostic criteria proposed in 2007 (1) eliminated the additional criteria as they are not essential for diagnosis; (2) recommended that plasma volume expansion should be performed with albumin rather than saline; (3) excluded creatinine clearance as it is impractical and difficult to interpret in chronic liver disease; (4) suggested that renal failure in the setting of ongoing bacterial infection should be considered as HRS, emphasizing that effective therapy should be started before the resolution of bacterial infection4).
Management
1. Hyponatremia
Restriction of free water intake is the mainstay of treatment for hyponatremia. Severe symptomatic hyponatremia should be managed with hypertonic saline infusion. However, both treatments are not aimed to address the underlying pathophysiology of water retention, the nonosmotic AVP secretion.
Pharmacologic inhibition of AVP action is emerging as a valuable addition to the armamentarium for management of hypervolemic or euvolemic hyponatremia. Binding to V2 receptor on the basolateral surface of the collecting tubular epithelial cells, V2 receptor antagonists (vaptans) inhibit the action of AVP and increase free water clearance. Several studies on cirrhotic patients with ascites and hyponatremia showed that V2 receptor antagonists increased urinary excretion of free water and plasma sodium concentration and were not associated with increased rates of serious adverse events33-35). A multi-center, randomized, placebo-controlled trial on 110 patients with ascites and hyponatremia reported that satavaptan improved the control of ascites, moderately increased plasma sodium concentration, and was not associated with significant alterations in plasma and urine electrolyte concentrations33). Patients who start on V2 receptor antagonists may require admission, as clinical trials with tolvaptan showed that a small fraction of patients (~2%) had early-stage increases in plasma sodium concentration higher than the acceptable range35).
2. Ascites
To counterbalance sodium retention in cirrhosis, negative sodium balance should be achieved by reducing daily sodium intake to less than 2 g/d and by starting diuretic therapy. Sodium restriction should be maintained throughout the entire course of ascites management. Mineralocorticoid antagonists are the first-line diuretics in management of ascites, as secondary hyperaldosteronism has been considered important in the pathogenesis of ascites. Loop diuretics and thiazide may be added if natriuresis is insufficient. As excessive natriuresis can greatly reduce intravascular volume and precipitate acute renal failure, caution should be exercised especially for patients with ascites in the absence of peripheral edema. Refractory ascites should be managed with therapeutic paracentesis with albumin replacement, construction of peritoneovenous shunt, or transhepatic intrajugular portosystemic shunt (TIPS).
3. Hepatorenal syndrome
An ideal treatment for HRS should be aimed at improving renal perfusion and glomerular filtration rate. It should achieve (1) reduction of serum creatinine to less than 1.5 mg/dL, (2) prolongation of survival, and (3) avoidance of serious adverse effects. Despite remarkable advances in the understanding of the pathophysiology of HRS, no ideal medical therapy has been discovered so far.
Liver transplantation is the only treatment proven to prolong survival in patients with HRS. It is important, however, to provide adequate management aimed at reversal of azotemia, because preoperative azotemia negatively affects the prognosis of the patients who underwent liver transplantation, and because effective treatment for HRS can earn valuable time for patients who await liver transplantation3).
Given the central role of splanchnic vasodilatation and hyperdynamic circulation in the development of renal dysfunction, new medical therapies are designed to reduce intense arterial vasodilatation and to expand the effective circulating volume. Systemic vasoconstrictor therapies, combined with albumin infusion, were found to be effective in patients with HRS in reversing renal dysfunction, and the survival of the patients who recovered from their renal dysfunction was significantly improved36-39). The available vasoconstrictors proven to be effective in clinical trials are vasopressin analogues (e.g., terlipressin, ornipressin)36,37), midodrine plus octreotide38), and norepinephrine39). Complications such as myocardial infarction, cerebrovascular disease or hypertension may preclude the use of vasoconstrictors. Concomitant volume expansion with intravenous albumin is essential, as vasoconstrictor therapy without albumin produced little effect in reversing azotemia40). An international group of experts issued treatment recommendations for the management of type 1 HRS4).
TIPS may be considered as a therapeutic option for HRS, especially when the risk of hepatic encephalopathy is low. Unfortunately, most patients with HRS are too ill to undergo TIPS. The Model for End-stage Liver Disease (MELD) score, a scoring system developed to predict the survival of patients with chronic liver disease and to prioritize organ allocation to patients in more critical condition, can be used to predict the survival of patients who underwent TIPS. A computational model suggested that TIPS should not be recommended for patients with a MELD score >18, as their median survival would be less than 3 months41).
Renal replacement therapies should be considered when patients are awaiting a liver transplant or when there is the possibility of improvement in liver function. In one retrospective study, 30 percent of patients who required dialysis survived to liver transplantation42). Patients with an acute and potentially reversible liver dysfunction may benefit from renal replacement therapy until they recover their liver function. Management of HRS is summarized in Table 2.
4. Prevention of HRS
Two studies have evaluated the efficacy of preventive measures for HRS. Sort et al. reported that intravenous albumin infusion combined with antibiotics for patients with SBP was associated with reduced rate of impaired renal function and increased survival, as compared with antibiotics alone43). Intravascular volume expansion might have been helpful to reverse the progression of renal dysfunction in patients with SBP, as bacterial translocation and increased inflammatory cytokines might augment nitric oxide production through the putative tetrahydrobiopterin synthesis pathway.
A randomized trial reported significant benefits with the administration of norfloxacin at 400 mg/day to patients with cirrhosis who met the following criteria: (a) ascites fluid total protein <1.5 g/dL and either of the following: (b) a Child-Pugh score ≥9 points and serum bilirubin ≥3 mg/dL; or (c) serum creatinine ≥1.2 mg/dL, blood urea nitrogen ≥25 mg/dL, or serum sodium ≤130 mEq/L44). Norfloxacin was associated with decreased one-year probability of SBP and HRS, and improved 3-month survival.

 XML Download
XML Download