

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Primary Sjögren's syndrome (pSS) is a chronic, slowly progressive autoimmune disease characterized by xerostomia and dry eyes1). It predominantly affects women and occurs in all ages. About one-third of patients with Sjögren's syndrome show extraglandular involvements and are manifested with Raynaud's phenomenon, vasculitis, lymphoma, and various organ involvements, including the kidneys1). The renal involvement in pSS includes tubulointerstitial nephritis (TIN) causing tubular dysfunction and less commonly glomerulonephritis. Herein, we present the cases of two patients with Sjögren's syndrome whose initial manifestations were fracture and muscle weakness, respectively.
Go to : 
Case Reports
Case 1
A 60-year-old woman presented to the emergency department with left hip pain after falling down. The simple X-ray revealed transverse subtrochanteric fracture of the left femur with displacement of the distal part. The patient had been on a monthly risedronate for osteoporosis. Otherwise, she had been well until this event. She was admitted and underwent open reduction and internal fixation on the following day. The computed tomography demonstrated another incomplete subtrochanteric fracture line on the medial side of the right femur. In addition, low bone mineral density (T-score, -2.7) and multiple rib fractures were noted on dual-energy X-ray absorptiometry and bone scan, respectively.
Oddly, the urine dipstick test revealed a strong positivity for glucose at the serum glucose level of 126mg/dL. The blood chemistry and arterial blood gas analysis showed hypokalemia, hyperchloremic metabolic acidosis, hypophosphatemia, hypouricemia, and elevated alkaline phosphatase (ALP) level with a modestly decreased kidney function (Table 1). The urine anion gap, fractional excretion or daily excretion of several electrolytes, and transtubular potassium gradient are shown in Table 1: positive urine anion gap in the setting of normal anion gap metabolic acidosis, normoglycemic glucosuria, enhanced potassium secretion leading to hypokalemia, and renal wasting of phosphate. These findings were consistent with Fanconi syndrome with renal tubular acidosis (RTA). Moreover, her serum sodium level increased up to 151mmol/L on the sixth day of admission, although 0.9% normal saline or hypotonic fluid was administered. The urine osmolality was inappropriately low(190mOsm/kg), despite adequate plasma antidiuretic hormone secretion to increased serum osmolality (304 mOsm/kg); such a finding reflected a urine concentration defect. Potassium citrate powder was administered daily with intravenous potassium phosphate replacement.
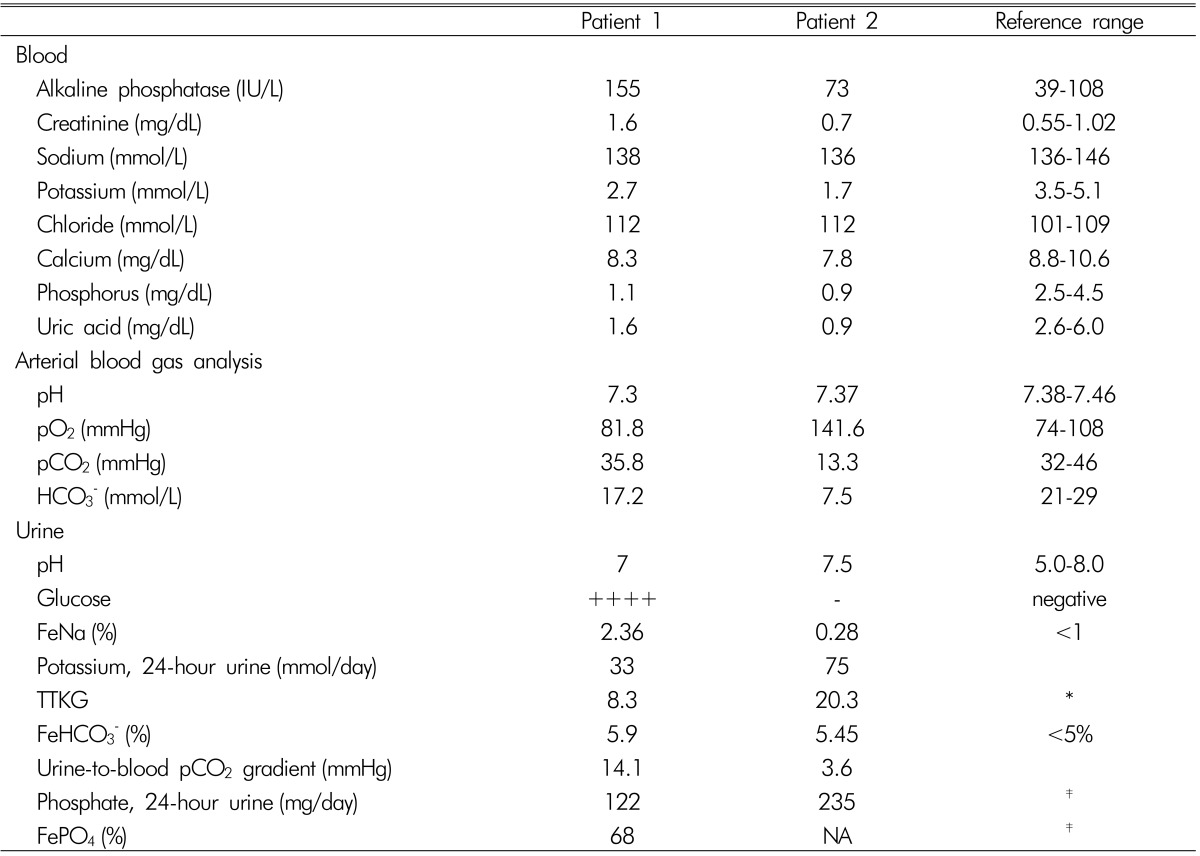
Table 1
Laboratory findings of our patients
FeNa, FeHCO3-, and FePO4 denote the fraction excretion of Na, HCO3-, and PO4, respectively; TTKG: transtubular potassium gradient; N/A: not available.
*In the setting of hypokalemia, the 24-hour urine potassium excretion level and TTKG should be less than 15-20mmol/day and 3-4, respectively.
†If the plasma HCO3 level is higher than 23-25mEq/L, the urine-to-blood pCO2 gradient should be higher than 20mmHg.
‡In the setting of hypophosphatemia, the 24-hour urine phosphate excretion level and FePO4 level should be <100mg/day and <5%, respectively.
![]()
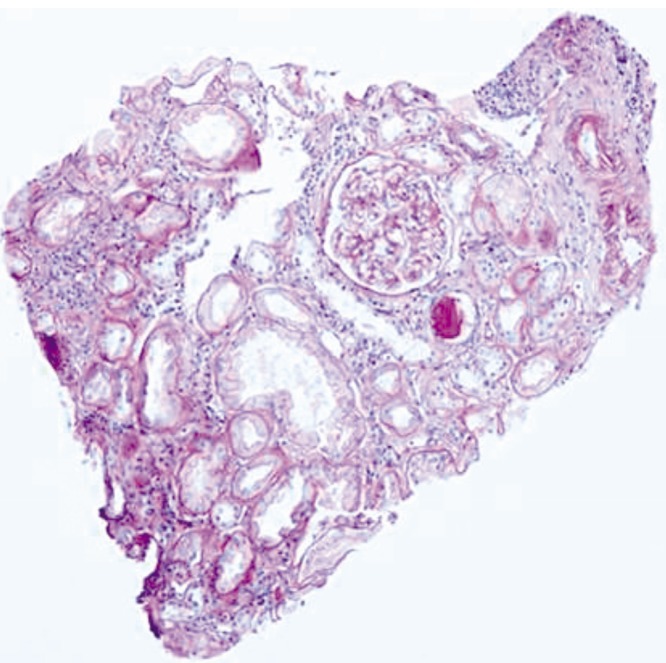
A renal biopsy was performed to determine the cause of chronic kidney disease and associated renal tubule dysfunction. The light microscopy showed a mild degree of interstitial nephritis and focal acute tubular injury with normal glomerulus (Fig. 1). However, the immunofluorescence and electron microscopy did not reveal any abnormal finding.
Further investigations revealed a speckled pattern of antinuclear antibodies (ANA) (1:160) with positivity for both anti-SS-A and anti-SS-B antibodies. She had a history of gritty sensation in the eyes and decreased tear flow on the Schirmer's test. Based on the clinical history and laboratory findings, interstitial nephritis associated with Fanconi syndrome, RTA, and nephrogenic diabetes insipidus secondary to Sjögren's syndrome were diagnosed. It was likely that RTA and hypophosphatemia were responsible for the multiple fractures, suggesting osteomalacia overlapping with osteoporosis.
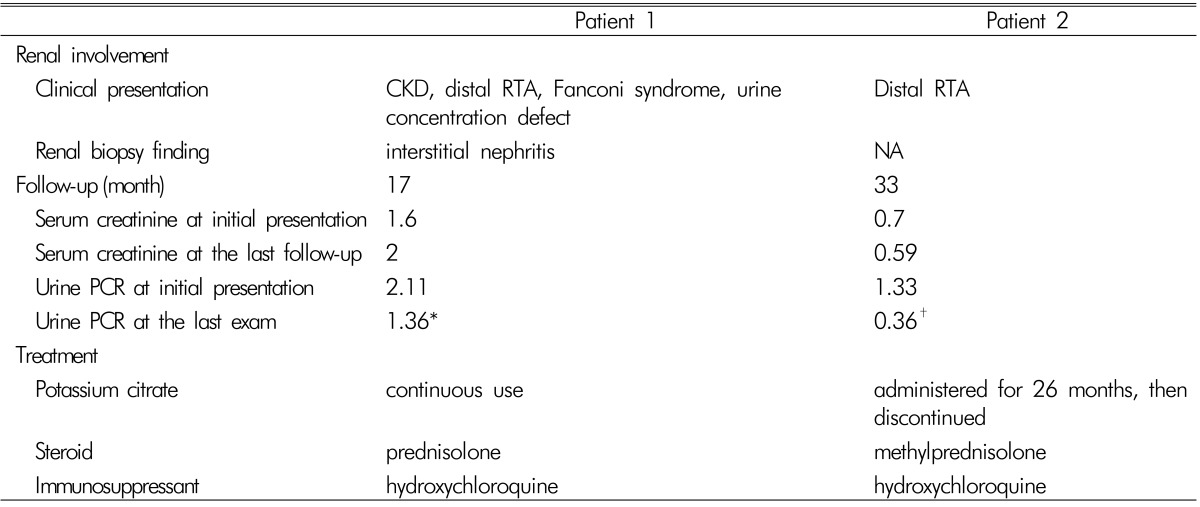
When the total carbon dioxide (CO2) level increased to 24.7mmol/L via the administration of potassium citrate, the fractional excretion of bicarbonate and urine-to-blood CO2 tension gradient (U-B pCO2) were calculated as <15% and <20mmHg, respectively, indicating distal RTA. Furthermore, her urine pH level persistently ranged from 6.5 to 8.0, which supported this type of RTA (Table 1). Thereafter, she has been on prednisolone and hydroxychloroquine with potassium citrate powder and maintained normal serum potassium, phosphate, and total CO2 level. The ALP level also decreased to the normal range. However, the serum creatinine level increased to 2.0 mg/dL after 17 months (Table 2).
Case 2
A 19-year-old girl was admitted with fever and weakness of both lower extremities. She was diagnosed with acute bronchitis and treated with intravenous levofloxacin. On admission, very low levels of serum potassium and phosphate were noted and thought to cause weakness on both lower extremities and rhabdomyolysis. In addition, hyperchloremic metabolic acidosis was accompanied. An excess urinary potassium loss and increased transtubular potassium gradient indicated excessive renal potassium excretion (Table 1). Moreover, an inappropriately high phosphate excretion in the presence of hypophosphatemia indicated renal phosphate wasting. Since hypokalemia of a renal origin and positive urine anion gap indicated the presence of RTA, the bicarbonate loading test was conducted to identify its specific type. Its results identified the diagnosis of distal RTA based on the decreased U-B pCO2 without excessive bicarbonate wasting (Table 1). Thereafter, sodium bicarbonate and potassium citrate were administered orally. Her symptoms resolved as the serum potassium and phosphate levels increased to the normal range.
Further evaluations for the etiology of RTA identified positive ANA(1:640, speckled nucleolar pattern) with positive anti-Ro and anti-La antibodies. However, she reported no dry eyes or mouth. In addition, the salivary scan and Schirmer's test did not show decreased salivary and tear flow. It was concluded that her motor weakness was attributed to distal RTA secondary to Sjögren's syndrome. Then, she has been treated with immunosuppressive agents of methylprednisolone and hydroxychloroquine and does not show any electrolyte imbalance and acidosis even after the discontinuation of alkali and potassium replacement (Table 2).
Go to :
Discussion
pSS can be manifested in different ways beyond dry mouth and eyes, sometimes even before sicca symptoms are developed. The renal involvement in pSS is a well-known extraglandular manifestation with a prevalence of 5-14% in most European studies23). In most cases, it affects the renal tubules through TIN and occasionally autoantibodies against a certain transporter. All segments of the nephron can be involved in pSS leading to distal and proximal RTA, Fanconi syndrome, diabetes insipidus, and less commonly Gitelman's syndrome and Bartter's syndrome4). Of these, distal RTA is the most frequent tubular dysfunction in pSS. The hypokalemia and metabolic acidosis in distal RTA cause muscle weakness, periodic paralysis5), and osteomalacia. In case of Fanconi syndrome, hypophosphatemia due to defective renal phosphate reabsorption is an additional contributing factor for osteomalacia.
Most RTAs in pSS are associated with TIN, which resembles lymphocyte infiltration in the exocrine glands. In these cases, the decreased expression of both H+-ATPase and anion exchanger 1 (AE1) in α-intercalated cells has been well observed without the presence of autoantibodies against those transporters678). However, underlying cellular events linking TIN and the absence of those two transport proteins have yet to be discovered.
There are previous reports of osteomalacia secondary to pSS leading to bone fractures9). The use of bisphosphonate in patient 1 could be responsible for the atypical left femoral fracture. However, according to the 2013 revised definition of atypical femur fractures10), incomplete fractures originating from the medial side of the subtrochanteric region were not consistent with atypical femoral fractures in which the fracture line starts from the lateral cortex. The looser zone on the right femur and multiple rib fractures with biochemical abnormalities of hypophosphatemia and elevated ALP levels suggest the presence of osteomalacia. A previous investigation reported that osteomalacia in pSS is a consequence of chronic metabolic acidosis and hypophosphatemia rather than pSS itself11). In a recent review of literature regarding previously reported cases of osteomalacia in pSS, the most common symptoms were bone pain and muscle weakness (85.3%) followed by fracture and pseudofractures (44.1%), and the most frequent laboratory findings were elevated ALP levels (79.4%) followed by reduced calcium and phosphate levels (70.6%)12).
Patient 1 appeared to have a long duration of disease, which was enough to cause softening of the bones. In contrast, patient 2 was diagnosed presumably at the early course of Sjögren's syndrome, whose manifestation was only muscle weakness. Although our two cases did not meet the pSS classification criteria proposed by the American-European Consensus Group13), pSS was a reasonable clinical diagnosis in the context of positive autoantibodies and its above-mentioned characteristic renal involvement. Moreover, the pSS classification criteria were not developed for use in clinical practice.
A course of glucocorticoid is the most widely used treatment option for patients with TIN secondary to pSS. It may prevent the development of interstitial fibrosis and tubular atrophy and resolve tubular dysfunction as well as the sicca symptoms as in patient 2. Maripuri et al. reported that prednisolone alone or with immunosuppressants, such as hydroxychloroquine, cyclophosphamide, or rituximab, improved the renal function of patients with pSS who had TIN on biopsy14). Although our patients were treated with the combination of corticosteroid and hydroxychloroquine, the efficacy of immunosuppressants as steroid-sparing strategies remains unknown in this setting, since clinical trials are difficult to conduct owing to the paucity of cases.
The involvement of multiple segments of the tubules in pSS as in patient 1 is an unusual phenomenon. She had a decreased renal function and lymphocyte interstitial infiltration reflecting moderate renal disease activity according to the EULAR Sjögren's syndrome Disease Activity Index3). Unfortunately, the disease had progressed until the development of fractures. If she had been diagnosed earlier, such an event would have been prevented and renal function preserved with appropriate treatment. In patient 2, the immunosuppressive agent induced clinical remission, which was enough to discontinue alkali and potassium replacement with marked reduction of proteinuria (Table 2). It is a challenge for physicians to make a diagnosis when encountering a patient with pSS without sicca symptoms. Since a prompt diagnosis may prevent serious complications and enable achievement of remission, it is of importance that Sjögren's syndrome should be considered in an unknown origin of hypokalemia and hyperchloremic metabolic acidosis.
Go to :
 XML Download
XML Download