

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Idiopathic demyelinating polyneuropathies are not common in the context of acute kidney injury (AKI). POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) is a rare disease characterized by the presence of proliferation of plasma cells with mainly lambda-chain restriction and diverse systemic symptoms occurring over several years, meaning that patients typically visit several clinical departments before obtaining the final diagnosis1). Kidney dysfunction is not included in the diagnostic criteria of POEMS syndrome, but AKI can be present with other systemic symptoms2). We report a patient with POEMS syndrome with recurrent AKI.
Case Report
A 52-year-old Korean man was admitted to this hospital because of abdominal distension and diarrhea. He had been well until 4 years ago, when he was diagnosed with cerebral infarct at another hospital. He was a businessman who rarely drank alcohol and had no previous history of medication. When he was transferred to our hospital 2 years ago, he had the symptom of mild tingling sensation in the left hand. One and a half years ago, paresthesia of right arm and intermittent difficulties with handwriting developed. Brain magnetic resonance imaging (MRI) with MR angiography showed neither infarct nor vascular stenosis. A diagnosis of transient ischemic attack was made. His renal function was normal (serum creatinine, Scr 0.88mg/dL).
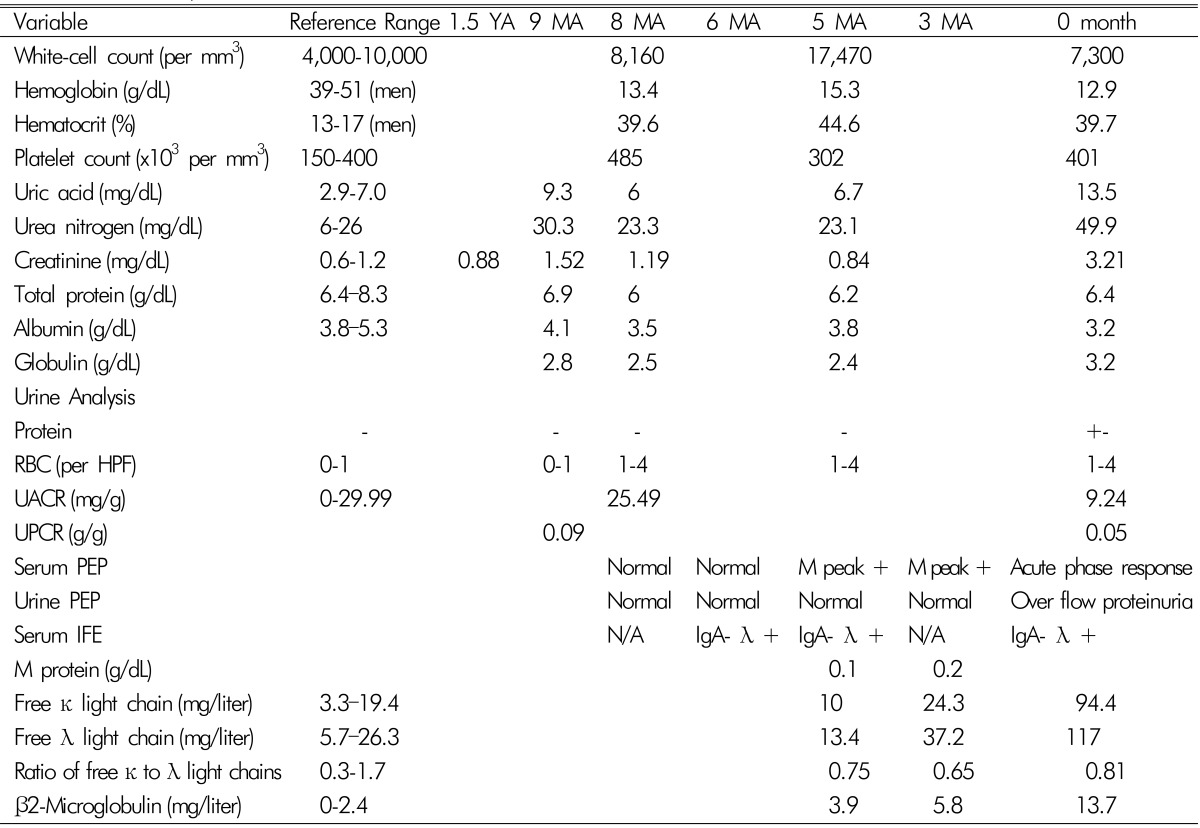
One year ago, he reported a new development of paresthesia of both feet. Nine months ago, after undergoing health checkups, he was referred to the nephrology, endocrinology, and ophthalmology clinics because of high Scr (1.52mg/dL), elevated thyroid stimulating hormone, and papilledema in both eyes, respectively. Urine analysis was normal and fractional excretion of sodium(FENa) was 0.16%. Since then, he suffered from gradually increasing paresthesia of both feet, progressing to both ankles and to both lower legs. He did not feel muscle weakness, but had some difficulties maintaining balance while walking as well as morning stiffness of both arms. Nerve conduction studies showed absent bilateral tibial nerve responses, slow motor responses on bilateral peroneal nerves, decreased sensory responses on bilateral sural nerves on the lower extremities, and slow motor and sensory nerve conduction velocities over the right median nerve. Electromyography reported no specific findings. On examination, his blood pressure was 100/60mmHg and no lymph nodes were palpable. Bilateral pitting edema was noted in both lower extremities. On cutaneous examination, red papules with diameters of 2 to 6mm, which developed in the previous year, were scattered on the truncal areas. Hypertrichosis with hyperpigmentation was observed in both hands and lower arms and finger clubbing was present. The pathologic diagnosis of the papules was glomerular hemangioma. Computed tomography (CT) of the chest and abdomen was performed to exclude malignancy. CT of abdomen showed multiple small lymph node enlargements around the aorta as well as a small amount of ascites. A diagnosis of chronic inflammatory demyelinating polyneuropathy was made and therapy with oral prednisolone was initiated. During the next 2 months, weakness in the patient's legs increased and he developed a limping gait. He could not walk on his toes and heels and his perception of vibration decreased in both knees. Nerve conduction studies showed deterioration compared with the previous study. The patient was treated with intravenous methylprednisolone pulse therapy. The test results for syphilis, hepatitis B and C, and HIV were negative. Tests for autoantibodies such as rheumatoid factor, anti-nuclear antibody, anti-ganglioside antibody, and anti-myelin-associated glycoprotein antibody, were all negative. Tests for vasculitis and anti-phospholipid syndrome were also negative. The levels of serum complements C3, C4 and immunoglobulins Ig G, A, and M were within normal limits. Cryoglobulin was negative. Additional laboratory results are shown in Table 1. A small M-peak was noted (0.1 g/dL) in serum protein electrophoresis (PEP) and no light chain was detected in urine PEP. Serum immunofixation electrophoresis (IFE) revealed findings of IgA-λ type monoclonal gammopathy.
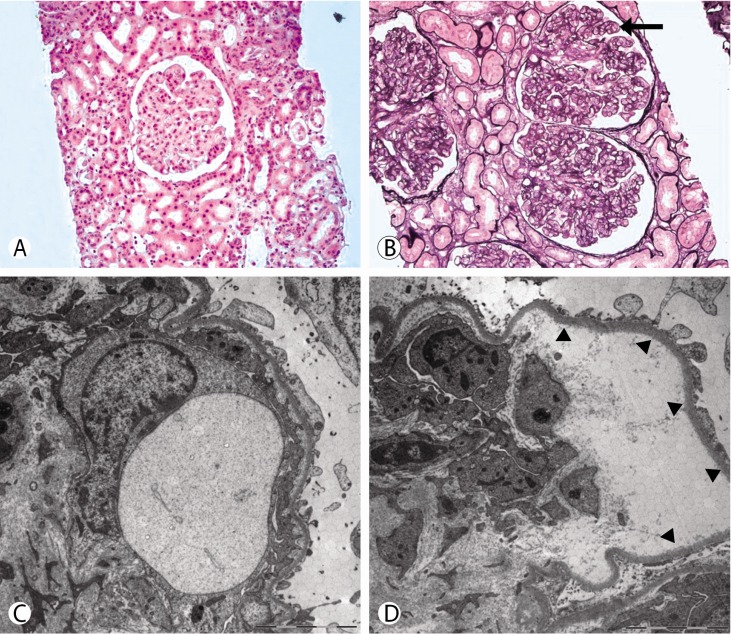
Six months before admission, he was referred to the oncologist for this finding and monoclonal gammopathy of undetermined significance (MGUS) was a plausible diagnosis. Bone marrow biopsy was deferred because he had taken steroid pulse therapy. Three months after steroid pulse therapy, his muscle weakness and gait improved, but burning paresthesia persisted. After 4 months, the steroid dose had been tapered and stopped. After stopping steroid treatment, he noted rapid progression of muscle weakness (he could not stand from the floor) and difficulties with walking and balancing. Although his appetite markedly decreased, his body weight increased by 5 kg and leg edema and abdominal girth increased. He began to have diarrhea. He had not taken any nephrotoxic agents including nonsteroidal anti-inflammatory drugs (NSAIDS). On examination, blood pressure was 120/80mmHg and there was bilateral foot drop. He reported difficulty breathing when he was supine in addition to a cough. He also suffered from running watery diarrhea, which leaked with coughing. Liver was palpable by two fingers. Laboratory results showed an increase in Scr to 3.21mg/dL and FENa and FEUrea was 0.24% and 12.3%, respectively. As abdominal distension increased with ascites he needed therapeutic paracentesis every 4 to 5 days. Ultrasonography showed normal-sized kidneys without hydronephrosis or distal ureter dilatation. Renal biopsy was performed to clarify the cause of AKI (Fig. 1). Renal pathology showed membranoproliferative glomerulonephritis (MPGN)-like lesions and mesangiolysis with capillary loops.
Light microscopic examination revealed renal cortex and medulla containing about 33 glomeruli, one of which was globally sclerotic. Most glomeruli showed increase in size and cellularity with mild to moderate endocapillary proliferations with diffuse double contours of capillary walls. There were diffuse interstitial edema and mild interstitial lymphocytes infiltrations with focal tubular atrophy containing hyaline casts destructing of tubular cells. Interlobular artery and arterioles showed swollen endothelial cells. Immunofluorescence study did not show any immunoglobulin or complement deposition or staining for kappa and lambda light chain. Electron microscopic examination revealed that most glomerular capillary lumina were compromised with diffuse duplications of capillary walls and subendothelial widenings with flocculent materials and mesangial interposition. Glomerular capillary endothelial cells are swollen with obliterated fenestrae. There were no electron dense deposits at any locations. Visceral epithelial cells showed hyperplasia and focal effacement of foot process about 20% of external capillary surfaces.
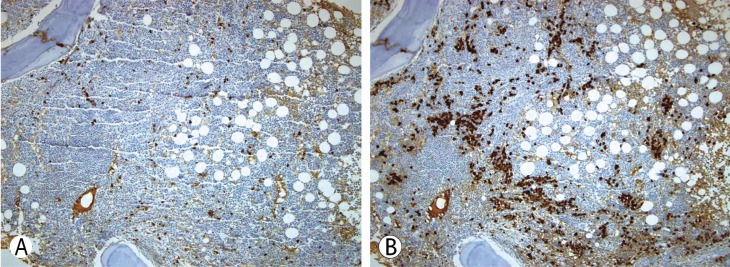
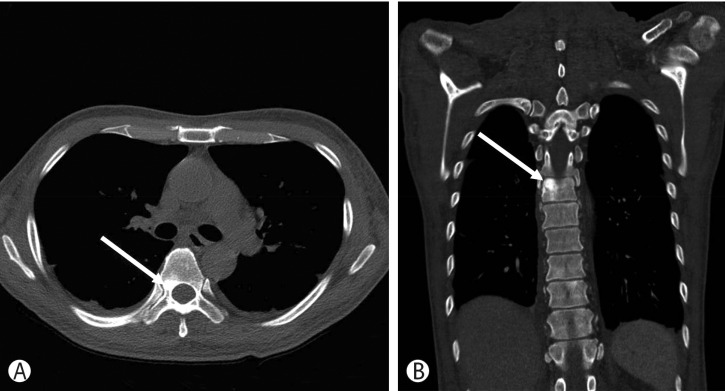
Bone marrow biopsy showed normal bone marrow cellularity (60%) and a normal ratio of the plasma cells (2.3%). Megakaryocyte hyperplasia was observed and a lymphoid follicle was seen in the bone marrow biopsy. Immunohistochemical staining showed that the majority of plasma cells were positive for lambda light chain (Fig. 2). The patient's diagnosis was almost certainly POEMS syndrome accompanied by recurrent AKI; however, an additional major criterion was required. Upon retrospective review of his previous chest and abdomen CT, we found osteosclerotic bone lesions in thoracic vertebral bones in the chest CT scan (Fig. 3).
Therefore, POEMS syndrome was diagnosed based on polyneuropathy, monoclonal plasma cell disorder with IgA lambda, sclerotic bone lesion, extravascular volume overload, endocrinopathy, hepatomegaly and skin changes, consistent with proposed diagnostic criteria. Once the diagnosis of POEMS had been established, the patient was treated with a 28 days cycle of bortezomib and dexamethasone combination followed by autologous hematopoietic stem cell transplantation (HSCT) at another hospital. After completion of chemotherapy, his symptoms improved gradually and the dialysis was stopped. His renal function and urine output improved as well. His serum creatinine reduced to 1.17mg/dL. Two months after the procedure, the patient showed a notable clinical improvement including polyneuropathy, normal Scr (0.80mg/dL) and mobilization was possible with the aid of a walker. The patient was still on rehabilitation therapy.
Discussion
POEMS syndrome is a rare paraneoplastic syndrome resulting from a proliferative monoclonal plasma cell disorder1). The diagnosis of POEMS syndrome must include two mandatory major criteria of (1) demyelinating polyneuropathy and (2) monoclonal plasma cell-proliferation (almost always λ type). In addition, one other major criterion must be present (any one of the following three features: osteosclerotic bone lesions [almost always present], elevated levels of vascular endothelial growth factor [VEGF], or Castleman disease). In addition, at least one of the following seven minor criteria should be met: organomegaly, extravascular volume overload, endocrinopathy (excluding diabetes mellitus or hypothyroidism), typical skin changes, papilledema, or thrombocytosis, polycythemia1). Making a diagnosis can be a challenge as the syndrome is rare and can be mistaken for other neurologic disorders.
The most prominent symptom of this patient was his progressive sensory neuropathy, followed by the development of motor weakness over a period of 4 years. His renal manifestation was recurrent AKI. From the nephrologist's perspective, possible differential diagnoses of polyneuropathy and kidney injury include nutritional deficiency such as pyridoxine or thiamine, human immunodeficiency virus-related neuropathy, amyloid, vasculitis, and paraneoplastic syndrome3). His laboratory results for viruses and vasculitis revealed no positive findings and his AKI had no definite cause. During the clinical course of this patient, there was no event of renal ischemia caused by nephrotoxic drugs or other systemic hemodynamic change. In the kidney, continuous endothelial injury caused mesangial interposition, mesangial proliferation and finally mesangial sclerosis. In addition to the glomerular change, ischemic damage caused by obliteration of arterioles and small arteries was proposed to induce recurrent acute kidney injury2).
VEGF is a potent microvascular permeability-enhancing mediator and a selective mitogen for vascular endothelial cell8). Therefore, the VEGF produced by plasma cells leads to a decrease in intravascular volume by increasing vascular permeability, which may contribute to prerenal AKI5).
We might discern from this case that serum and urine PEP should be obtained in patients with polyneuropathy and unexplained kidney injury to detect monoclonal gammopathy. However, serum and urine PEP of this patient were normal 8 and 6 months ago (Table 1). In POEMS syndrome, the median percent of plasma cells in the bone marrow is less than 5%, which is reflected by the small amount of M-protein, and serum PEP often cannot detect the M-peak1). Instead, serum and urine IFE should be performed to identify the isotype, as these techniques are three times more sensitive in detecting monoclonal immunoglobulin than serum PEP4).
In kidney biopsy, immunofluorescence staining showed no deposition of λ-light chain and electron microscopy showed no electron-dense deposits. These are quite different findings from other plasma cell disorders that showed kidney damage with deposition of a monoclonal immunoglobulin4). Instead, renal pathology of this patient showed extensive endothelial cell injury and evidence of increased extravascular permeability. Secretion of VEGF from the monoclonal plasma cells in patients with POEMS syndrome has been suspected to trigger these changes25), but the mechanism remains unknown and must be investigated6). In contrast to the pathologic lesions, proteinuria was absent and microscopic hematuria was minimal in this patient, which may reflect a relatively intact glomerular basement membrane structure and podocyte functions.
Treatment of POEMS syndrome is directed to target the underlying monoclonal plasma cell disease7) and therapeutic strategies for the treatment of multiple myeloma have been reported to show successful patient and renal outcomes and an improvement in morbidities related to treatment67). As this patient showed bone marrow involvement, he received systemic chemotherapy and had autologous blood stem cell transplantation.
 XML Download
XML Download