

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Isolated glycosuria is a non-disease condition characterized by increased urinary glucose excretion but a normal serum glucose level. Familial renal glycosuria (FRG) is an inherited renal tubular disorder in which there is persistent isolated glycosuria in the absence of hyperglycemia. It is caused by mutations in SLC5A2, the gene encoding the sodium glucose co-transporter (SGLT2). SGLT2 inhibition increases the renal excretion of glucose and sodium, thereby lowering the serum glucose level and reducing the plasma volume1). Although enhanced sodium excretion is thought to contribute to the reduction in plasma volume and lowering of the blood pressure (BP), clinical data on urinary sodium excretion in FRG are limited2). There have also been no studies on the role of fasting and postprandial changes in urinary sodium excretion in patients with FRG. Here, we report a case of isolated renal glycosuria in a healthy young man with an SLC5A2 gene mutation. In this patient, we investigated the change in the urinary Na+/glucose concentration after glucose loading to evaluate the tubular glucose and sodium reabsorption capacity in FRG.
CASE REPORT
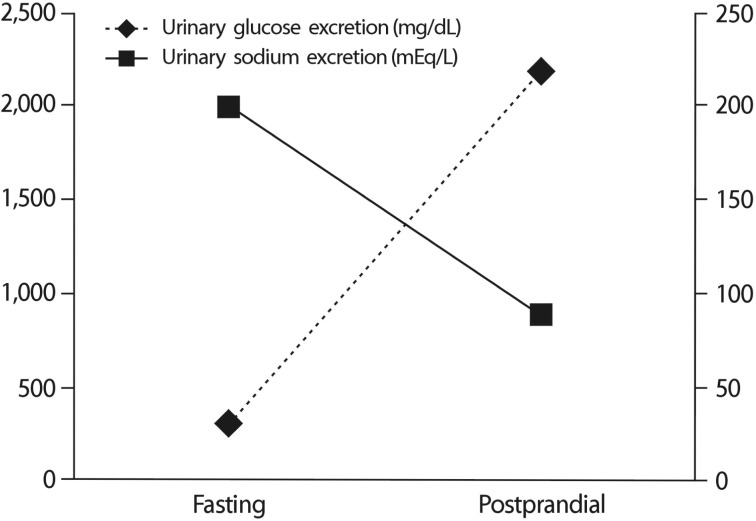
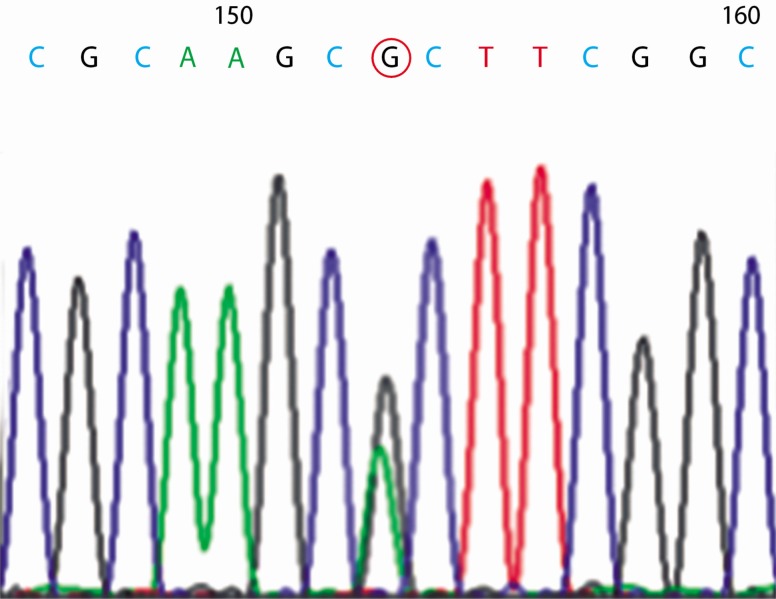
A 26-year-old man visited our outpatient clinic for the evaluation of incidentally found glycosuria. The patient was a healthy military officer in whom glycosuria, but no other abnormal lab findings, was detected on repeated routine screening. He had no manifestation of renal disease and was not taking any medication. His mother had the same history of incidental glycosuria on routine screening. Neither an examination of his physiological systems nor physical examination revealed any abnormal finding. His laboratory results did not indicate tubular dysfunction, except renal glycosuria, or any other condition that could contribute to his hyperglycemia. His fasting blood glucose level was 84mg/dL, PP2 glucose level was 126mg/dL, and HbA1C level was 5.4%. His blood urea nitrogen and creatinine levels were 13.3 and 1.21mg/dL, respectively. Dipstick urinalysis showed 4+ glucose, pH 5.0; however, blood and protein were absent. His 24-h urinary glucose level was 3,700mg, and creatinine excretion level was 1.71g/day. An examination of the patient for fasting and postprandial changes in urinary glucose and electrolytes showed fasting spot urinary glucose level of 295mg/dL and PP2 urinary glucose level of 2,170mg/dL. Fasting and postprandial urinary sodium excretion levels were 200mEq/L and 89mEq/L, respectively (Fig. 1). Fasting and postprandial urinary osmolarities were 902mOsm/kg and 834 mOsm/kg, respectively. Sequencing of the patient's SLC5A2 gene showed a heterozygous missense mutation of c.395 G>A in exon 4 that resulted in the replacement of an arginine with a histidine at position 132 (p.R132H) of the protein (Fig. 2).
Discussion
Under normal physiological conditions, the kidney reabsorbs all of the filtered glucose via the proximal tubule, in a process mediated by SGLT2, a kidney-specific, low-affinity/high-capacity Na+/glucose co-transporter. A defect in this transporter causes glycosuria3). The SLC5A2 gene encodes SGLT2, and mutations in SLC5A2 are responsible for renal glycosuria, and it is commonly associated with FRG4). The first report of an SLC5A2 mutation in FRG was published in 20004). Subsequently, another study with a larger number of patients confirmed that SLC5A2 mutations are responsible for the vast majority of FRG cases5). Our patient was heterozygous for the missense mutation c.395 G>A in exon 4, causing the substitution of histidine for arginine at position 132 (p.R132H) in the protein. This allele has been previously reported6).
FRG is an inherited renal tubular disorder characterized by persistent isolated glycosuria in the absence of hyperglycemia. The safe and normal lives of patients with FRG accelerated the development of SGLT2 inhibitors. An emerging new class of oral antidiabetic drugs, SGLT2 inhibitors, significantly reduced not only HbA1C level but also systolic BP in obese patients with type 2 diabetes mellitus. One suggested mechanism is that SGLT2 inhibitors decrease BP via osmotic diuresis induced by urinary glucose and sodium excretion and loss of body weight1). Our patient had a fasting urinary sodium excretion level of 200mEq/L; however, his postprandial urinary sodium excretion level was 89mEq/L. There is no previous report of fasting and postprandial differences in the urinary sodium concentration of patients with FRG. We think that the mechanism underlying this difference may be a compensatory one, protecting against osmotic diuresis induced by the increased excretion of glucose in the urine. It likely involves an increase in the sodium reuptake at the loop of Henle and at more distal tubules. However, further studies on both urinary sodium and glucose excretion in patients with FRG are required.
 XML Download
XML Download