

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Gitelman's syndrome (GS) is the most common inherited renal tubular disorder1). GS results from the defect of Na-Cl cotransporter (NCC) on distal convoluted tubules. It expresses hypokalemia, hypomagnesemia, and hypochloremic metabolic alkalosis2). It has a long asymptomatic period and often discovered incidentally after adolescence compared to Bartter's syndrome. The cause of the defect of NCC is the mutation on the SLC12A3 gene, and it has been reported in more than 400 GS-related mutations. Herein, we report on a case of GS, diagnosed by renal biopsy and genetic analysis which present missense mutation in nucleotide 179, cytosine to threonine (c.179C >T) in Exon 1 of SLC12A3).
Go to : 
Case Report
A 42-year-old man presenting an anterior chest pain, visited the emergency room. According to his medical history, although he visited the hospital with a chest pain repeatedly, heart work up, like electrocardiography and cardiac muscle enzymes, showed non-specific findings and a trans-thoracic echocardiogram revealed normal heart function. He did not take any drugs or herbal medications. On physical examination, blood pressure was 110/60mmHg and pulse rate was 82/min and regular. There was no pretibial edema or other neurologic abnormalities. The results of the blood test showed: sodium 140mEq/L, potassium 2.7mEq/L, chloride 91mEq/L, total calcium 9.3mg/dL (9.5-10.9), ionized calcium 2.09mmol/L(2.3-2.58), magnesium 1.1mg/dL (1.9-2.5), ionized magnesium 0.26mmol/L (0.45-0.6) and trans-tubular potasium gradient (TTKG) 12.68. Arterial blood gas analysis showed: pH 7.459, pCO2 42.4mmHg, pO2 88.5mmHg, HCO3 29.7mEq/L, and SaO2 97.3%. The results of urinalysis showed: sodium 80mEq/L, potassium 28.2mEq/L, chloride 148mEq/L, calcium 2.3mg/dL, creatinine 21.7mg/dL, spot urine Ca/Cr 0.1, and 24 hours urine magnesium 21.0mEq/L/day (5-17). Laboratory test showed hypochloremic metabolic alkalosis with hypokalemia, hypomagnesemia and hypocalciuria. Chest X-ray and kidney ultrasonography showed nonpathognomonic findings.
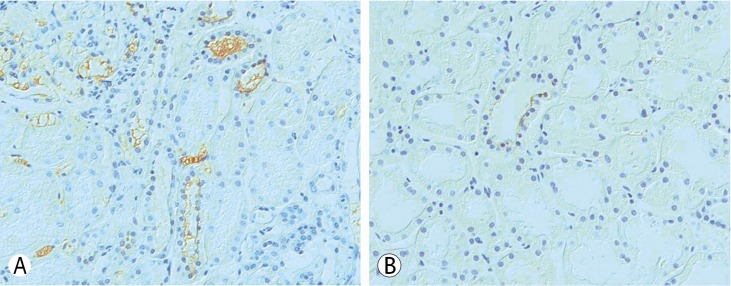
To evaluate for hypokalemia, which persists over 10 years combined with other electrolyte imbalances, we took an ultrasonography guided kidney biopsy. There were 3 global sclerosises among 51 glomeruli, and the light microscopy finding was normal without minimal increase of mesangial cell proliferation. Immunofluorescence findings showed all negativity for IgG, IgA, IgM, C3, C4, C1q, and lambda in the glomerulus. The result of the immunohistochemistry of NCC on tubules, and the expression degree was decreased compared with the control (Fig. 1).
We performed genetic analysis for diagnosis of GS. Although his family (parents and 3 sisters) did not show any clinical abnormality or electrolyte imbalances, we analyzed genomic DNA of the patient and his family in Keimyung University Medical Genetics Institute. Genomic DNA was isolated from peripheral blood by using the Wizard Genomic DNA Purification Kit according to the manufacture's instruction (Promega, Madison, WI, USA). Amplification of the coding sequence of the SLC12A3 gene by polymerase chain reaction was carried out as previously described. Polymerase chain reaction-single strand conformational polymorphism (PCR-SSCP) was performed. PCR products in the patient, his family, and normal control were electrophoresed through 10% polyacrylamide gels. The result in patient, a double mutant band was found, his family had only one band (Fig. 2).
 | Fig. 2The polyacrylamide gel electrophoresis of the SLC12A3 gene SSCP profiles in Gitelman's syndrome. A homozygous mutation revealed two of new line (arrow) and heterozygous mutation revealed only one. The patient is diagnosed as GS and his family is carrier of GS. Abbreviations: N; normal control, P; patient, F1; father, F2; mother, F3; old sister, F4; first younger sister, F5; second younger sister, SSCP; single strand conformational polymorphism, GS; Gitelman's syndrome.
|
To get apparent result of sequencing, PCR products were cloned by TOPOTA cloning (Invitrogen Ltd., Paisley, United Kingdom) and direct sequencing was performed on those PCR products that showed altered band mobility in the above analysis. Variations in the sequences between GS and matched normal sample were performed using the ABI 3730 DNA sequencer (Bionics Inc, Korea). The result revealed that the patient had homozygous missense mutation of nucleotide 179, cytosine to threonine (ACG to ATG) on exon 1 of the SLC12A3 gene. His family has heterozygous mutation at the same region, and supports the diagnosis of GS and carrier (Figs. 3, 4).
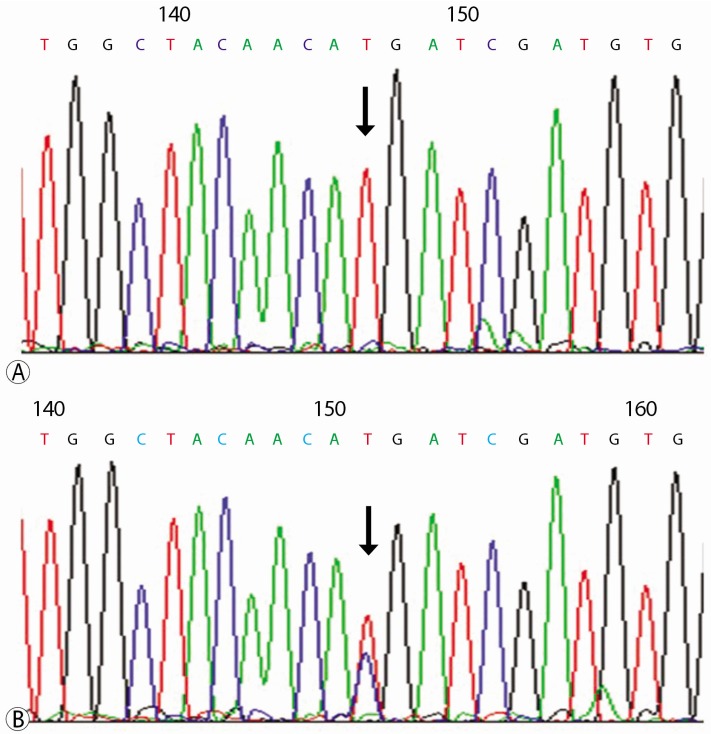 | Fig. 3(A) DNA sequence of SLC12A3 gene presents homozygous mutation of c.179C>T in patient, (B) and heterozygous mutation of c.179C>T in patient's family.
|
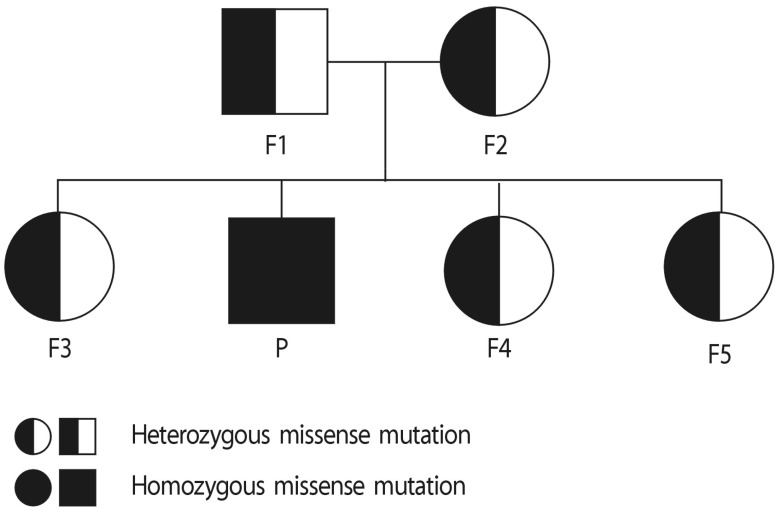 | Fig. 4A pedigree analysis presented patient's parents and his 3 sisters were carrier too. The potassium level of all carriers were over 3.5mEq/L and they didn't have any clinical symptoms. Abbreviations: P, patient; F1, father; F2, mother; F3, old sister; F4, first younger sister; F5, second younger sister; GS, Gitelman's syndrome.
|
Go to :
Discussion
Hypokalemia is a common finding in overall patients, furthermore it occurs in more than 20% of hospitalized patients4). The kidney is a key regulator that controls the homeostasis of potassium, 80% of potassium transit the kidney. Therefore, it needs to be considered about renal tubular dysfunction in patients with hypokalemia. Especially, if adult patients who have other electrolyte imbalance, like hypomagnesemia and hypocalciuria, in addition to hypokalemia, come to the hospital. We have to distinguish GS from Bartter's syndrome.
GS was first reported in 1966 by Gitelman. It is a thiazide-like salt-losing tubulopathy which shows familial hypokalemic and hypochloremic metabolic alkalosis5). It has developed by a mutation of SLC12A3 gene that encodes thiazide-sensitive NCC on distal convoluted tubule. SLC12A3 gene is located in chromosome 16q13, which has 26 exons2). GS related SLC12A3 mutations have been reported to have more than 400 different forms, from missense mutation to gross deletion and insertion mutation3). We report a case of homozygous missense mutation in c.179C>T on exon 1 that leads transposition of codon 60, threonine to methionine. The c.179C>T mutation has been commonly detected in Asia including Japan and China6789). This is the first report which analyzes pedigree with c.179C>T mutation in Korea.
A carrier state of SLC12A3 gene mutation was reported in 1% of the general population. Although GS is the most common inherited tubular disorder, it was not commonly diagnosed in the clinical field1). The clinical manifestation of GS is diverse from being symptomless to tetany, growth retardation and fatal cardiac arrhythmia. GS is common to be found incidentally with mild symptoms. This case was diagnosed by an attentive inspection of the electrolyte imbalance, even though the symptom was not specific. After being diagnosed as GS, he took oral magnesium and potassium and he did not complain about the chest discomfort and weakness anymore.
Actually, homozygous mutations in SLC12A3 gene are only reported in 18% of patients while heterozygous mutations are more frequently observed1011). But, the type and character of the mutation does not clearly correlate with the symptoms12). The family of the patient has heterozygous mutation in SLC12A3 gene. Their values of electrolytes, such as sodium, potassium, and chloride were normal and they did not complain of any symptoms. Although we did not evaluate other serum and urine electrolytes, and chemistry of the family, they need to be evaluated even if they experience nonspecific symptoms. Knowledge about the accurate location and pattern of the gene mutation can lead us to better understand the function of SLC12A3 gene and the disease entity of GS. It provides not only early diagnosis, but also the direction of improvement. If physicians suspect GS, they have to do a genetic analysis which is a gold standard tool for diagnosing GS. And, this positive attitude leads to early detection and improves the quality of life of the patient with GS presenting non-specific symptom like as fatigue, thirst, dizziness, general weakness.
Go to :
 XML Download
XML Download