

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Familial renal hypouricemia is caused by a defect in renal tubular urate transport. The disorder is majorly caused by mutations in the gene SLC22A12, coding for human urate transporter 1 (URAT1)1). Familial renal hypouricemia patients are usually asymptomatic, but some may form renal stones or be predisposed to acute renal failure2). We present a case of familial renal hypouricemia with a history of recurrent exercise-induced acute kidney injury (EIAKI). The diagnosis was confirmed by genotyping of SLC22A12.
Case Presentation
1. First episode
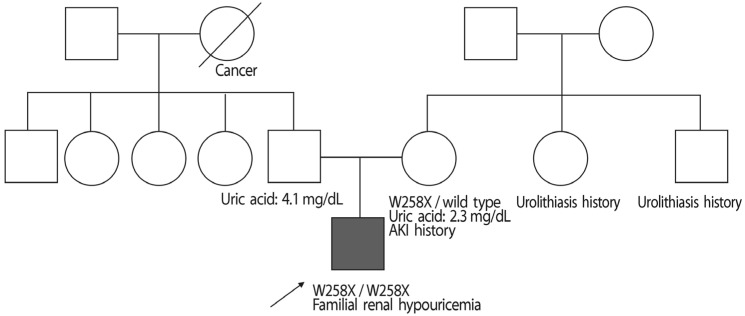
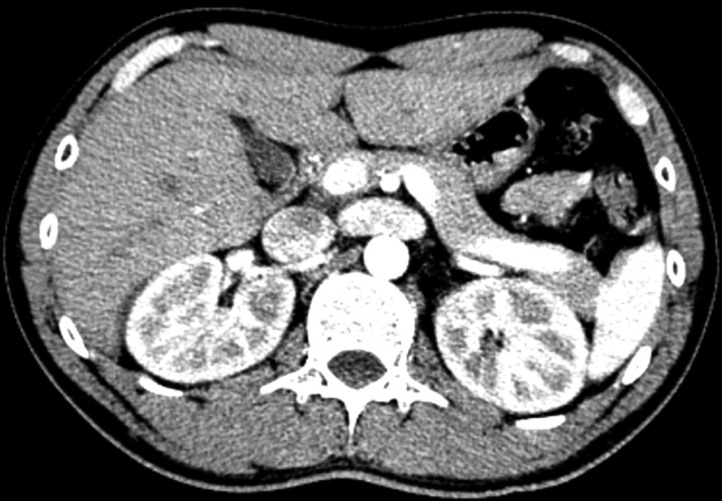
A 24-year-old Korean male visited the emergency room (ER) of our hospital on April 1, 2015 with pain in both flanks. The initial impression was ureter stone due to his symptom; thus, a computerized tomography (CT) of his abdomen was performed. Abdomen CT showed a decreased enhancement of both kidneys but no evidence of urolithiasis. After ruling out urolithiasis, the patient was admitted for diagnostic evaluation. Upon history taking, he stated that he had suffered from urolithiasis 4 years prior, and had passed the stone spontaneously. He also stated that his mother had a history of AKI, and both his aunt and uncle, had a history of urolithiasis. Despite the patient's mother's AKI history, no other specific medical history was noted with familial history taking (Fig. 1). His blood pressure was 130/70mmHg, and his weight and height were 78 kg and 170 cm, respectively. The patient showed pain in both flanks. Laboratory tests on admission revealed the following results: Cr, 1.6mg/dL; blood urea nitrogen (BUN), 17mg/dL; C-reactive protein, 1.40 mg/dL; white blood cell count, 11,380/µL; potassium, 4.0 mmol/L; lactate dehydrogenase, 494 U/L (normal range: 263-450 U/L), creatine kinase, 86 U/L (normal range; 60- 220 U/L); total CO2, 24.8mmol/L; and a low uric acid level of 0.5mg/dL. The urinalysis revealed a specific gravity of 1.012, a pH of 5.0, and 0-1 red blood cells/µL. No glycosuria, aminoaciduria was found. The 24-hour urinary protein was 17mg. The following parameters were within normal limits or negative: serum immunoglobulin; serum complement; antinuclear antibody; perinuclear/ cytosolic-antineutrophil cytoplasmic antibody; and HIV, HBsAg, and hepatitis C antibodies. Fractional excretion of uric acid (FeUA) was 23.95%. Abdominal ultrasonography was unremarkable. After conventional causes of Cr elevation were ruled out, the patient's history was taken again, during which he stated that he had been swimming prior to the flank pain event. The patient was suspected to have familial renal hypouricemia based on, the exclusion of other causes of hypouricemia, familial histories of AKI and urolithiasis, and decreased renal enhancement on abdominal CT (Fig. 2). Thus, the patient was also suspected to be in an EIAKI state. Therefore, genetic analysis for SLC22A12 mutations was conducted. The patient was discharged 6 days after admission. During the outpatient follow up, the patient was stable and the Cr level was maintained at <1.0mg/dL.
2. Second episode
AKI recurred 10 weeks after the first episode. On June 17, 2015, the patient was admitted to our hospital via the ER due to pain in both flanks that occurred following swimming. The physical examination did not reveal abnormalities. The patient's blood pressure was 120/80 mmHg, his 24-hour urine protein excretion was 718mg. In addition, serum Cr was 2.2mg/dL, BUN was 21mg/dL, and uric acid was 2.4mg/dL. The renal ultrasound showed increased echogenicity of both kidneys, but structural abnormalities were not observed. We again performed a FeUA and the result was 14.61%. The history of flank pain after swimming may indicate recurrent EIAKI. Hydration and symptomatic treatment were performed during the admission. The symptoms of the patient resolved upon treatment and the serum Cr level decreased gradually. The patient was discharged on day 9 of hospitalization. Anaerobic exertion was prohibited including swimming. At the time of the 1-month follow up, the patient had a normal serum Cr level of 1.0mg/dL, but his hypouricemia (uric acid, 0.7mg/dL) remained. No further episode of EIAKI was noted.
3. Molecular biologic analysis
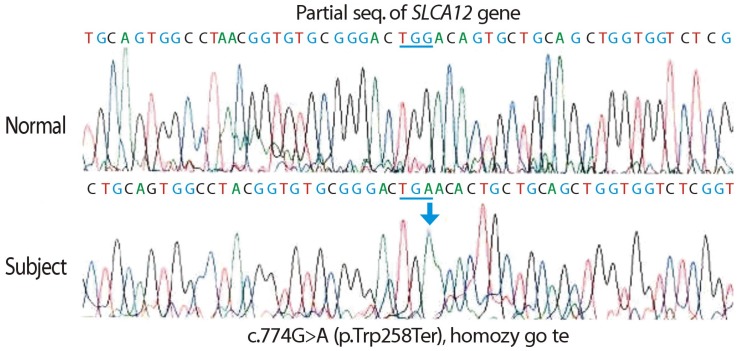
Sequence analysis of the SLC22A12 gene was performed on the patient and his mother due to the suspicion of EIAKI associated with familial renal hypouricemia. T he sequence analysis of SLC22A12 revealed a mutation of c.774 G>A (p.[Trp258Ter]:[Trp258Ter]), or homozygous W258X, which causes renal hypouricemia. The patient's mother exhibited a heterozygous mutation of W258X, making her a carrier of renal hypouricemia; her results were consistent with autosomal recessive inheritance of the disease (Fig. 3). We also proposed another molecular biologic analysis to the patient's father, but the father refused further genotyping since he had normal uric acid level and the cost.
Discussion
Familial renal hypouricemia is a rare hereditary renal disorder specific for uric acid. The defective urate reabsorption in the proximal tubules results in the increased renal urate clearance. It is distinguished from other hereditary conditions, such as Fanconi and Hartnup syndromes, in which the urate transport defect is only one component of a generalized disturbance of membrane transport1). The disease is characterized by a low uric acid level and predisposition to EIAKI and urolithiasis. CT imaging of familial renal hypouricemia patients is reported3) to show decreased enhancement, which is explained by transient vasoconstriction or constriction of the renal tubules.
A recent molecular biologic study established that urate is reabsorbed via URAT1 on the tubular apical membrane, and that mutations in SLC22A12, which encodes URAT1, cause familial renal hypouricemia4). Mutational alteration of SLC22A12 in familial renal hypouricemia was first identified by Enomoto et al. in 200215). On the other hand, Vitart et al. (2008) reported that mutations of SLC2A9, which encodes a high-capacity urate transporter (GLUT9), could also cause familial renal hypouricemia 67).
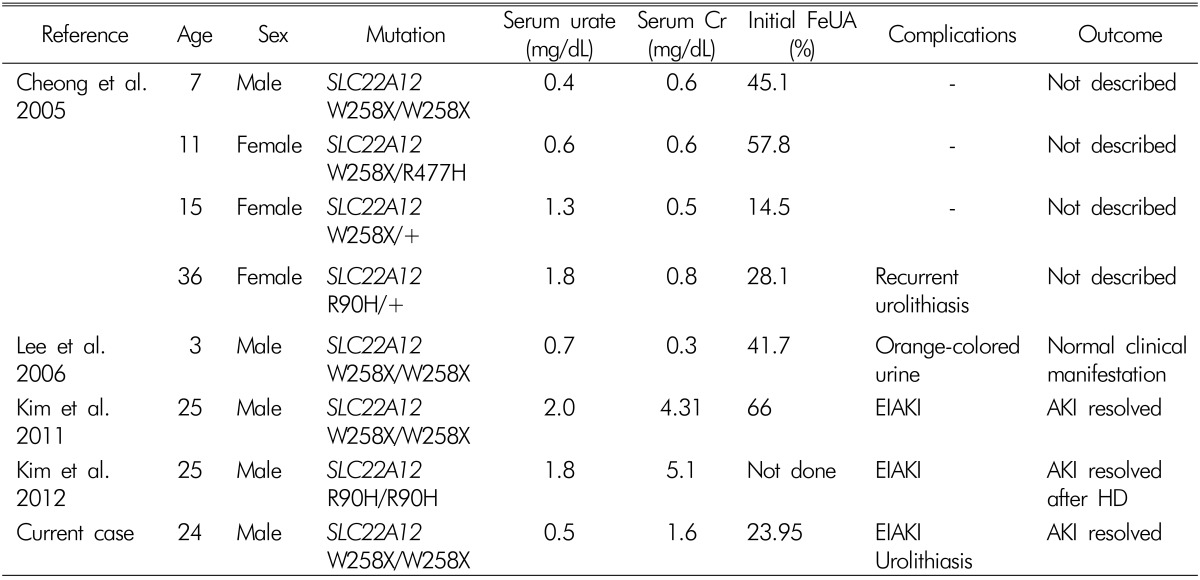
Familial renal hypouricemia has been reported to be a rare disease entity; to the best of our knowledge only seven cases in which a genetic mutation was confirmed have been reported in Korea (Table 1). Two cases89) published in 2011 and 2012 featured EIAKI; one case10) in 2004 featured recurrent urolithiasis, and another case published in 2006 featured orange-colored urine. Three cases showed a W258X homozygous mutation 81011), two showed a W258X heterozygous mutation (including W258X/R477H allele), one9) showed a R90H homozygous mutation, and another10) showed a R90H heterozygous mutation. In Japan and Korea to date9), the predominant mutation of SLC22A12 in familial renal hypouricemia cases is W258X. The case presented herein also exhibited this mutation.
The essential pathogenetic cause of familial renal hypouricemia is associated with proximal tubule urate transporters. URAT1 is expressed on the luminal membrane of proximal tubular cells and operates as an exchanger, taking up urate from the primary urine in exchange for intracellular nicotinate or lactate121314).
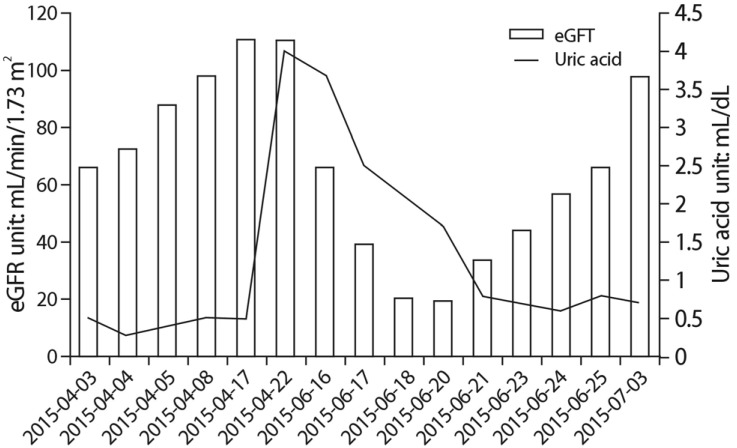
Although most familial renal hypouricemia patients are clinically asymptomatic and detected incidentally, some may have nephrolithiasis or be predisposed to EIAKI, as in the case presented herein. The mechanism of EIAKI remains unclear, but nephrologists have proposed several possible explanations. One explanation emphasizes the role of uric acid as an antioxidant. Anaerobic exercise results in accumulation of oxygen free radicals, which have a vasoconstrictive effect in the kidney. According to these explanations, familial renal hypouricemia patients have decreased renal antioxidant potential, which might lead to kidney injury caused by reactive oxygen species15). There were other mechanisms suggested, as post exercise uric acid elevation from skeletal muscle origin16), and AKI followed by uric acid crystal precipitation at renal tubules15), but further evidence must be established. During the episodes, the patient exhibited AKI following the exertion. Considering the patient's hypouricemia and the genotyping, the Cr elevation might be interpreted as EIAKI. The patient's renal profile and symptoms were improved by treatment during hospitalization and outpatient follow-up (Fig. 4).
In our case, the patient showed features of hypouricemia and EIAKI; thus, a diagnosis of familial hypouricemia was established. Based on this, genotyping was performed, revealing a homozygous mutation of W258X. Genotyping of the patient's mother was also performed, which revealed that she carried a heterozygous mutation of W258X. Compared to the patient, the patient's mother showed a higher level of uric acid and a normal renal profile. The clinical features of EIAKI in the patient, and the genotyping results, confirmed the diagnosis of familial renal hypouricemia.
Conclusion
In conclusion, our patient had a history of EIAKI and current laboratory findings of hypouricemia; thus, genotyping of SLC22A12 was performed due to suspicion of familial renal hypouricemia. The diagnosis of familial renal hypouricemia was confirmed by the genotyping. He was prohibited from engaging in anaerobic exercises.
Familial renal hypouricemia is a rare disorder but must be considered in a patient with a low uric acid level, elevated Cr, familial history of urolithiasis and EIAKI. SLC22A12 mutation screening is an important molecular investigation for the diagnosis of familial renal hypourhypouricemia, and should be conducted in patients suspected to have the disease.
 XML Download
XML Download