

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
For a long time, to survive, the human body has adapted to lack of nutrition including energy, salt, and water. With excess nutrition, the burden of obesity is a growing problem worldwide although malnutrition still remains unsolved in undeveloped areas. The problem is the imbalance between energy intake and expenditure that leads to various human disorders. Many scientists have studied the mechanisms of the energy balance which help them understand variable disorders of cells, tissues, organs and the whole body. All of endocrine system, body energy stores and central nervous system interact together for an efficient energy balance of the human body (Fig. 1). The reason why obesity-associated hypertension is increasingly recognized as an important phenotype is that both obesity and hypertension are components of the metabolic syndrome (including type 2 diabetes mellitus and dyslipidemia). These are all major risk factors for cardiovascular disease. The consequence of obesity is an alarming increase in adults, even in children. Otherwise, obesity increases the risk for type 2 diabetes mellitus, cardiovascular disease, cancers, and musculoskeletal disorder, and pulmonary disease1). Although obesity is an important risk factor for diabetes and hypertension, the two most common etiologies of chronic kidney disease, obesity itself may potentiate kidney injury. In many researches, independent of kidney injury, obesity is also associated with the development of hypertension. However, the mechanisms underlying obesity-associated hypertension have not yet been clearly understood. In this review, we briefly summarize several mechanisms that provide the understandings of obesity-associated hypertension.
Go to : 
Putative mechanisms of obesity-associated hypertension
There are a large number of hypothetical mechanisms via which obesity is able to cause hypertension (Fig. 2)2). They include the renin-angiotensin-aldosterone system (RAAS), the sympathetic nervous system(SNS), metabolic dysregulation (including hyperinsulinemia, adipokine imbalance, and increased inflammatory cytokines).
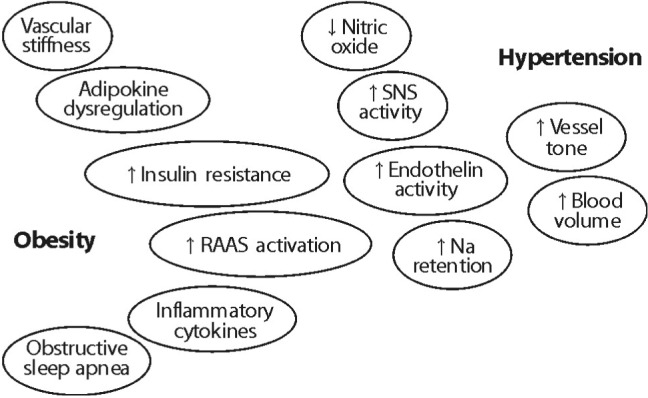
Activation of the RAAS is a key contributing factor in obesity3). Angiotensin II, a potent vasoconstrictor and aldosterone with other RAAS metabolites have been known to underlie obesity-associated hypertension. Several mechanisms (including the SNS stimulation of renin release) have been considered to cause RAAS activation. Recent studies have reported the RAAS activation in adipose tissue, especially visceral adipocytes with the generation of angiotensin II and aldosterone.
Hyperinsulinemia in obesity is a possible factor in obesity-associated hypertension. Insulin induces the activation of the SNS2). Insulin also has a direct action on the kidney to stimulate sodium retention. As adipokine, leptin produced in adipocytes, suppresses appetite potently and stimulates the SNS. Leptin deficiency causes severe obesity in animals and humans. Both insulin-mediated and leptin-mediated SNS activation may be considered as mechanisms compensated in obese patients to stabilize weight and restore energy balance by stimulating sympathetically mediated thermogenesis with consequent enhancement of energy expenditure2).
Another contributing factor in obesity-associated hypertension is vascular stiffness which is influenced by adipokines such as leptin and hyperinsulinemia in obesity4). There are also oxidative stress and inflammation that leads to endothelial dysfunction. Decreased nitric oxide impairs vascular relaxation leading to vasoconstriction for hypertension.
There are three major factors among the leading hypotheses regarding the underlying mechanisms responsible for obesity-associated hypertension. They are adipose tissue dysfunction, activation of the RAAS, and endothelial dysfunction.
Go to :
Adipose tissue dysfunction: Obesity-induced inflammation
Excess energy-induced fat accumulation contributes to the harmful disorder of many tissues, such as the pancreas, liver, skeletal muscle, heart, and vessels5). Multiple molecular mechanisms affect on the development of obesity-related complications. Recent data provides that the innate inflammatory activity might be an important role in the pathophysiology of obesity-related diseases6). In particular, the inflammatory cells of monocytes/macrophages are considered as an important contributing factor. Obesity is accompanied by infiltration and activation of macrophages in adipose tissue, leading to chronic inflammation of adipose tissue. Adipose tissue has been reported by Hotamisiligil et al. that it is an important organ of obesity-induced inflammation determined by expressing proinflammatory molecules7). The phenotypic changes of adipocytes occur into hypertrophy and an inflammatory response in an autocrine and paracrine manner to impair adipocyte function, including insulin signaling8). Recently the inflammatory molecules of adipose tissue for understanding these mechanisms have been recognized5-7). Adipose tissue has been considered as an important source of obesity-induced inflammation by the inflammatory molecules, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), and inducible nitric oxide (iNOS)6,7).
Despite adipose tissue not only secreting inflammatory mediators, but it also responds to inflammatory signals. They express various receptors for cytokines, chemokines, and growth factors. However, a contribution of adipocytes to secretion of proinflammatory adipokines is low in most instances. Interestingly in metabolic disorders, this low-grade inflammatory response in adipose tissue can occur persistently, which is different from that in autoimmune diseases. Recently, an inflammation in adipose tissue has been reported in the infiltration of immune cells such as monocytes/macrophages. Thus, the view for the role of adipose tissue has been extended into an immunologically-active organ9).
The molecular mechanisms underlying monocyte/macrophage recruitment to adipose tissue is not clearly understood. A recent review revealed that various stimuli, including lipopolysaccharide and free fatty acids, activate the intracellular transcriptional nuclear factor kappa B (NF-κB) pathway via TNF receptor 1, IL-6 receptor, and toll-like receptor (TLR)-4. The NF-κB pathway plays a bridging role in the innate immunity with metabolism9,10). Of note, recent studies observed that obesity induces adipose tissue inflammation infiltrated by distinct immune cells, especially macrophages and T lymphocytes11,12). Macrophages might be largely responsible for the enhanced inflammatory activity of adipose tissue in obesity. In addition, obesity changed the phenotype of macrophage in adipose tissue13). Macrophages might have a heterogenetic function in response to different stimuli. In the lean and insulin-sensitive milieu, macrophages are polarized toward M2 or "alternatively activated" macrophages. Excess nourished status induces hypertrophic changes in adipocytes secreting chemokines, that are differentiated into M1 or "classical" polarized macrophages. M1-polarized macrophages release TNF-α, IL-6, IL-10, and iNOS that cause activation of the NF-κB pathway in adipocytes14). Eventually the specific macrophage population of adipose tissue is involved in obesity-related adipose tissue inflammation, which is tightly linked to insulin resistance15,16).
Go to :
Adipokines and neuropeptides: A link between the adipose tissue and hypertension
Adipose tissue is a highly metabolic organ with pleuripotent functions far beyond the mere storage of energy. Adipose tissue produce and secrete several molecules such as leptin, resistin, adiponectin, and visfatin, as well as cytokines such as TNF-α, IL-6, MCP-1, and IL-1. These adipokines are stimulated via the intracellular signaling pathways that regulate inflammation of adipose tissue.
Leptin is a peptide hormone secreted from adipose tissue into circulation. It has effects on the hypothalamus to decrease appetite and stimulate sympathetic activity leading to induce hypertension. These actions are mediated by two pathways, which are through either the propiomelanocortin-derived peptides-αmelanocyte-stimulating hormone (MSH)-driven by high leptin levels, or agouti-related peptide and neuropeptide Y (NPY). MSH bind to melanocortin receptors to stimulate the SNS, elevate energy expenditure and activate the hypothalamus-pituitary-adrenal axis. Normally, NPY is suppressed by high leptin level, but, interestingly, NPY, overexpressed in the leptin-resistant state of obesity, rather activate the SNS leading to hypertension.
Other peptide such as adiponectin, ghrelin, and endocannabinoids are related with insulin resistance to be involved with obesity-associated hypertension.
Go to :
The RAAS and adipose tissue
Clinical trials for hypertension have presented the result of lower incidence of diabetes mellitus among patients treated with RAAS inhibitor than other classes of anti-hypertensive drugs17-19). Despite these studies were performed on patients with hypertension or congestive heart failure, a relative risk for new-onset diabetes mellitus was reduced in a meta-analysis of randomized controlled trials. Moreover, the activity of RAAS is also associated with the metabolic syndrome such as obesity20). Interestingly, mature adipocytes express all components of RAAS, including angiotensinogen, angiotensin-converting enzyme, as well as angiotensin type 1 (AT1) and angiotensin type 2 (AT2) receptors21). Angiotensinogen mRNA in murine and human adipocytes was identified in the late 1980s22). Angiotensinogen is produced predominantly from white adipose tissue and liver. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation23). Plasma angiotensinogen has been thought to be a reflection of hepatic production. However, Massiéra F et al. investigated whether adipose angiotensinogen is released locally and into the blood stream using transgenic mice with angiotensinogen expression restricted to adipose tissue. They demonstrated that angiotensinogen produced by adipose tissue has an influence on both the local tissue and systemic circulation, which supports a role of adipose angiotensinogen in obesity-associated hypertension. Consistently, angiotensinogen expression in adipose tissue of obesity of rats and humans were demonstrated24,25). Elsewhere, other RAAS components, including angiotensin converting enzyme, renin, renin receptor, AT1 receptor, AT2 receptor, and Mas receptor are produced from adipose tissue (Fig. 3)26-32). This data suggest a local tissue RAAS also works at adipocyte level. As a result, angiotensinogen is served into adipocyte hypertrophy and hypertension via angiotensin II, which induce systemic vasoconstriction, sodium and water retention and aldosterone production.
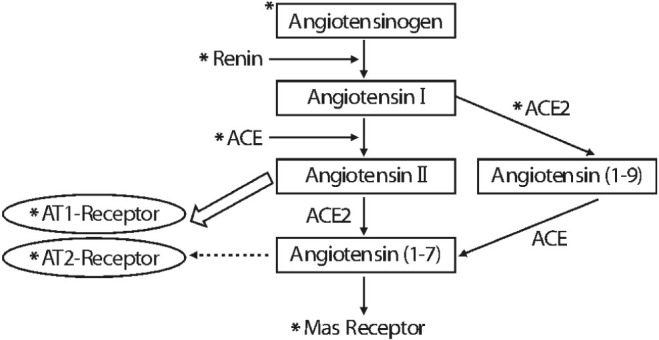
More interestingly, angiotensin II has a major role in promoting lipid storage in mature adipocytes. Yvan-Charvet and Quignard-Boulange demonstrated that angiotensin II increased cholesterol uptake into murine adipocytes via phosphatidylinositol 3 kinase (PI3K)-dependent translocation of scavenger receptor33). AT1 antagonist-treated mice revealed enhanced plasma free fatty acid and AT2-deficient mice showed low glucose uptake in adipose tissue. They suggest that angiotensin II functions as a lipogenic hormone through AT2 receptor in human and murine adipocyte. However, our study showed that angiotensin II decreased glucose uptake and lipid storage in adipocyte34). On the other hand, receptor antagonist of either AT1 or AT2 decreased glucose uptake and restored lipid contents in adipocyte. In addition, there is rare evidence for the action of angiotensin II on other receptors such as Mas receptor. Therefore, the exact role of angiotensin II and angiotensin receptor in the mechanisms of adipocyte metabolism, remains unclear.
Go to :
Microvascular dysfunction and insulin resistance
Endothelial dysfunction and arterial stiffness are considered an independent predictor for the progression of cardiovascular events and chronic kidney disease contributing to systolic hypertension35,36). Elderly people have a greater chance to progress into those changes, but young obese people present similar changes in them. Inflammation and oxidative stress in adipose tissue are important to interact with the microvascular endothelium in the mechanisms of obesity-associated hypertension37). Especially inflammation and oxidative stress in visceral perivascular adipose tissue are associated with augmented systemic inflammation and oxidative stress. Finally, they are strongly interrelated through a vicious cycle, leading to endothelial dysfunction and vascular hypertrophy, where microvascular endothelium-dependent vasodilatation is impaired in response to vasodilators such as insulin. Increased microvascular resistance raises blood pressure and decreases blood flow in organs including skeletal muscle, leading to metabolic disorder. Otherwise interestingly, various stimuli of free fatty acid, angiotensin II, proinflammatory molecules induce vascular insulin resistance. Insulin signaling pathways in vascular endothelial cell are based on the balance between vasoconstriction and vasodilatation37). Through insulin receptor, insulin receptor substrate-1 (IRS-1), phosphatidylinositol 3 kinase (PI3K), Akt, endothelial NO synthase (eNOS), and nitric oxide (NO) consist of vasodilatory pathway. On the other hand, the activation of extracellular signal-regulated kinase 1/2 (ERK 1/2), mitogen activated phosphate kinase (MAPK), and endothelin-1 (ET-1) lead to vasoconstriction. TNFα, angiotensin II, and free fatty acid produced from perivas- cular adipose tissue activate Akt pathway, but not ERK1/2 pathway. However, adiponectin stimulates AMPK and inhibit MAPK, leading to the activation of eNOS. Therefore, a regulatory link between microvascular and perivascular adipose tissue inflammation and adipokine synthesis are provided to explain the mechanism of obesity-associa- ted hypertension.
Go to :
Conclusion
Obesity has been implicated in increasing the risk for cardiovascular diseases. The complex interactions between obesity and hypertension still raise many questions regarding the exact mechanisms to prove cause and effect of them. Further research focusing on both adipokines and neuropeptides may provide simultaneously the new therapeutic target of obesity and hypertension (Fig. 4). A better understanding of the pathophysiology of obesity-associated hypertension may promise the possible self-control for appetite and blood pressure in the near future.
Go to :
 XML Download
XML Download