

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Camurati-Engelmann disease (CED), also known as progressive diaphyseal dysplasia, is an autosomal dominant disease characterized by apparently thin and long extremities, limb pain, muscle weakness, and radiographic findings of cortical thickening of the long bone diaphysis.12) It is a very rare disease, with a frequency estimated at 1 in 1,000,000.3) Mutations of transforming growth factor beta 1 (TGFB1) gene were identified as causative for this phenotype;4) however, some patients with the same phenotype do not have any mutation in that gene.5) CED is classified as type I (MIM 131300) when a pathogenic mutation in TGFB1 is identified; otherwise, it is classified as type II (MIM 606631).5)
The severity of clinical manifestations and radiological abnormalities and onset of disease are variable from patient to patient, which along with the rarity of this disease makes a timely diagnosis challenging. Especially in childhood, it may present only with gait disturbances and muscle weakness, misleading doctors to consider neuromuscular disorders. The purpose of this study was to investigate the mode of presentation of mutation-confirmed type I CED in order to facilitate adequate diagnostic workup.
METHODS
This is a retrospective study approved by the Institutional Review Board of Seoul National Univeristy Hospital (IRB No. H-1504-014-662). Written consents were obtained from all patients or their parents for presentation of the case or images. In total, 8 type I CED patients were recruited from our skeletal dysplasia database. Genetic study for TGFB1 gene mutation was performed by Sanger sequencing, and pathogenic mutation was confirmed (Table 1). They were 4 sporadic patients, and two pairs of mother and son. There were 6 males and 2 females. We conducted a thorough review of their medical records and radiographic data. The age of symptom onset, mode of initial presentation, and process of diagnostic work-up, followed by the cause of referral to the authors, causative mutation in the TGFB1 gene, and course of disease progress were recorded (Table 1). The bone segment(s) involved, their symmetry, and progression of bony involvement with age were reviewed on the available plain radiographs. Patients were classified into three groups according to the mode of initial presentation to medical service.
RESULTS
We categorized the patients into three groups according to the mode of presentation.
Group I comprised 4 patients presenting with muscle weakness or gait disturbance at a young age. The mean age at symptom onset was 3.8 years (range, 1.7 to 5 years), and that at the first hospital visit was 4.1 years (range, 2.3 to 5.5 years). Three of them (patients 1–3) were initially seen by orthopedists and then referred to pediatric neurologists, and the remaining 1 patient (patient 4) was first seen by a pediatrician. They underwent electromyography (EMG) examination and muscle biopsy, which showed a nonspecific myopathic pattern or no abnormality (Table 1). They had been followed under a tentative diagnosis of myopathy until they developed limb pain, and were referred to the authors at an average age of 16.8 years (range, 8.6 to 29 years). The severity of limb pain varied from being transient and subsiding without any intervention (patient 2) to making the subject wheelchair-bound (patient 4). Skeletal survey and subsequent molecular test for TGFB1 mutation confirmed the diagnosis of type I CED in these patients.
Group II consisted of three patients (patients 5, 6, and 7) whose main complaint was limb pain at the initial presentation. There were two mothers of the two patients in group I (patients 6 and 7). Their mean age at onset of symptom was 13.6 years (range, 9.9 to 16 years). The severity of limb pain ranged from mild (patient 5) that was tolerable without any medication to severe that required narcotic analgesics (patient 7). They finally visited the authors at age 20, 46, and 59 years, respectively, and the diagnosis of type I CED was made based on plain radiographic findings and genetic tests. The latest ambulatory status of these patients ranged from ambulatory without (patient 5) or with intermittent pain (patient 6) to wheelchair-bound (patient 7). One patient (patient 8) in group III was found to have multiple asymptomatic bony lesions on plain radiographs taken after a minor vehicle accident at age 8 years. He was followed under a tentative diagnosis of polyostotic fibrous dysplasia for 10 years. CED was not considered because of the lack of limb pain at that time. He recalled having mild intermittent leg pain in the morning or after sports activities, which he had not complained of until specifically asked. He was diagnosed with type I CED when presented to the authors at age 18 years. At the latest follow-up, he remained asymptomatic.
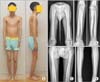
All patients in group I showed a slender body habitus with a body mass index (BMI) in the underweight range (Table 1). Two of 3 patients in group II and the only patient in group III showed a BMI in the normal range. None of the patients with a BMI in the normal range belonged to group I. There was no patient showing a BMI in the overweight range in our series.
Other musculoskeletal manifestations included flat feet (patient 2); bilateral radial head dislocation and bilateral ankle valgus deformity (patients 4 and 7); and bilateral hallux valgus with subluxation of the first metatarsophalangeal joints (patient 7).
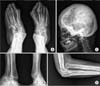
Skeletal survey of all patients revealed symmetrical cortical hyperostosis in various severities with or without expansion of medullary cavities in the long bones in various combination of involvement, including the skull (Figs. 1, 2, 3). Radionuclide bone scans of 3 patients (patients 4, 6, and 8) showed diffuse hot uptake in the affected areas of long bone segments.
DISCUSSION
In the present study, we described a variety of clinical and radiographic phenotypes of type I CED patients. We categorized them into three groups according to the mode of initial presentation because clinical features of the disease changed over time to resemble each other during the progression of the pathologic condition. We could not find any notable difference in the mode of manifestation according to the mutation.
Limb pain was the most common clinical symptom of CED, comprising 68% in a big series of CED.3) However, the rarity and subsequent unawareness of this disease made a timely diagnosis difficult as in patient 6 who underwent unnecessary bone biopsy and remained undiagnosed until age of 43 years. It appears that in young patients as those in group I of our series, muscle weakness and motor disturbance are the prevailing symptoms, which leads to the use of EMG and muscle biopsy and subsequent misdiagnosis of myopathy. Misdiagnosis of CED as myopathy has been reported in several studies with 678) or without910111213) confirmation of TGFB1 mutations. Muscle biopsy for identification of specific changes usually gives negative results,10) except for atrophy of muscle fibers14) as in our series. TGFB1 inhibits myogenesis15) and adipogenesis16) which might explain the prevalence of underweight in CED patients.3) BMI was in the underweight range for 5 patients of our series, whereas it was within the normal range in the remaining 3 patients. However, low BMI and slender body habitus are not always seen in CED patients. Some studies described 2 CED patients with obesity (BMI, 27 kg/m2) and limb pain.1718)
Although the majority of CED is diagnosed before the age of 30 years,319) the asymptomatic course of the disease might contribute to delayed diagnosis. Patient 8 was categorized into a separate group (group III) due to the absence of pain at the time of diagnosis, however he experienced pain for some period in adulthood. If the patient had not taken radiographs for a motor vehicle accident, he could have belonged to group II or remained undiagnosed. CED has been incidentally found in several patients with19) or without1120) confirmed TGFB1 mutation. Low et al.10) reported a 62-year-old lady without any limb pain, whose headache along with radiographic findings of the skull and long bones led to the diagnosis of CED.
Radiographic findings of CED include (1) hyperostosis of one or more of the long bones that begins in the diaphyses of the long bones and can progress to the metaphyses and rarely to the epiphyses; (2) periosteal involvement with uneven thickening and increased diameter of the cortices; (3) endosteal bony sclerosis that can lead to narrowed medullary canal; and (4) which are usually symmetric in the appendicular skeleton but may be asymmetric. 21) The femur (98%) and tibia (96%) are the most frequently and initially involved bone segments followed by the humerus (88%), radius (87%), and ulna (85%).22) Cortical thickening along the long bone diaphyses occurs as a result of both endosteal and periosteal bone formation, leading to diaphyseal broadening and narrowing of the medullary canals. Contrarily, endosteal bone removal appears to be defective and broadening of the medullary canal may ensue as a result.23) As the disease progress, the metaphyses also become affected and late involvement of the femoral capital epiphyses, albeit rare, has been reported. 1) In our series, the affected mother in a family revealed epiphyseal involvement of the femoral heads (patient 7). Involvement of the short tubular bones is uncommon.2) We had one patient only involving the metacarpals as thickened endosteum that was found incidentally by skeletal survey (patient 5). Other radiological findings variably seen are skull involvement beginning at the base of the anterior and middle fossae and often including the frontal bone, and mild osteosclerosis in the posterior neural arch of the spine and parts of the flat bones that correspond to the diaphysis.21) Scintigraphy could be normal in patients with apparent radiographic hallmarks as described by Clybouw et al.24) Thus, it is advised to combine radiographic examination with bone scintigraphy to confirm or rule out the diagnosis of CED.
In spite of the proposed conservative3,25) and operative19) treatment options, no effective treatment has been established for CED. Losartan was reported to be effective in relieving limb pain26) due to down-regulation of the expression of TGFB type 1 and 2 receptors.27) However, 3 patients in our series (patients 3, 4, and 6) did not respond to Losartan treatment. Patient 7 was prescribed oxycodone/ naloxone for pain control and showed borderline response. The remaining two patients did not require any medical intervention.
In summary, CED presents with a wide range of clinical manifestations including muscle weakness and motor disturbance in early childhood and limb pain in later childhood or adulthood, and it can be even asymptomatic. Awareness of this rare disease entity may be the key to timely, accurate diagnosis. This disease should be considered in the differential diagnosis of limb pain or motor disturbance in children to avoid unnecessary diagnostic work-up. Furthermore, physicians in other fields, such as pediatrics and neurology, should be well acquainted with possible clinical manifestations of this disease.


 XML Download
XML Download