

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Vogt-Koyanagi-Harada (VKH) disease is an idiopathic, multisystem autoimmune disorder characterized by its affects on pigmented tissues in the ocular, auditory, integumentary, and central nervous systems. The prevalence of VKH disease varies markedly, and the risk of developing at least one neurological manifestation exceeds 50%.1 Certain neurological manifestations-including aseptic meningitis, encephalitis, encephalomyelitis, and cranial nerve neuropathy-are occasionally associated with this disorder, but acute myelitis has rarely been reported. Early systemic administration of corticosteroids will suppress the acute inflammatory process, and prevent recurrences and the development of complications.
We present a case of VKH disease accompanied by acute myelitis, and review two previously published case reports in an attempt to elucidate the pathogenesis.
Case Report
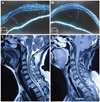
A 50-year-old Chinese Han woman presented with sudden onset of difficulty walking, numbness on the left side of the body, and difficulty with urination for 6 days. Twelve days prior to the presentation she had been diagnosed with incomplete VKH disease by an ophthalmologist based on blurred vision in both eyes, bilateral nontraumatic granulomatous iridocyclitis, retinal edema, and the presence of exudates. She had received corticosteroid treatment (500-mg intravenous methylprednisone for 3 days followed by 300-mg intravenous methylprednisone for 3 days and a tapering course of 80-mg prednisone for 6 days). Neurological manifestations (headache, tinnitus, difficulty climbing stairs, numbness on the left side of the body, and dysuria) emerged during the tapering of steroid treatment. She recalled a history of upper respiratory tract infection a month previously but denied any history of vaccination. Physical examinations revealed normal vital signs. There was no lymphadenopathy, or oral or genital ulcers. Her skin and hair showed no vitiligo, poliosis, or alopecia. Neurological examinations showed a normal mental status and cranial nerves except for a visual acuity of 20/200 bilaterally and papilledema of both eyes in a fundus examination. The strength of both upper and lower limbs was decreased. Her muscle tone was increased. Her deep tendon reflexes were hyperactive without clonus. The Babinski sign was present bilaterally, and her response to the finger-to-nose test was impaired. Her gait was slow and stiffly shuffling. Pinprick sensations were decreased in the left limbs, whereas position sense was preserved. No obvious sensory level was identified. Kernig's sign was absent. Routine laboratory evaluation showed all relevant values to be within normal limits. Screening for connective-tissue disease, including C reactive protein, rheumatic factor, antinuclear antibody, C-antineutrophil cytoplasmic autoantibody, anti-double-strand DNA, anti-Smith, anti-ribonucleoprotein, anti-Sjogren's syndrome A, and anti-Sjogren's syndrome B antibodies, produced normal or negative results. The erythrocyte sedimentation rate was 39 mm/hr. Cerebrospinal fluid (CSF) contained 128 WBCs/mm3 (normal <10/mm3) with 94% mononuclear cells and 4.83 g/L protein (normal 1.5-4.5 g/L). A CSF bacterial culture produced negative results. A nerve conduction study showed mild polyneuropathy, and visual evoked potentials were normal. A chest X-ray was normal. A brain MRI scan was normal with no callosal or significant periventricular lesions. Optical coherence tomography performed independently by two ophthalmologists revealed bilateral disc edema accompanied by serous retinal detachment (Fig. 1A and B). Spinal-cord MRI (Fig. 1C and D) revealed a hyperintense signal in the T2-weighted sequence with significant gadolinium enhancement between the C1-3 vertebrae in the cervical cord. The patient was diagnosed as aseptic meningitis with acute myelitis complicating VKH disease, and was given pulse steroid therapy again (intravenous administration of 1000 mg of methylprednisolone for 3 days, 500-mg intravenous methylprednisolone for 10 days, 250-mg intravenous methylprednisolone for 10 days, and a tapering course of 60-mg prednisone over a 6-month period). After 4 months she was able to climb stairs without help. Her visual acuity recovered to 50/200 and her numbness and dysuria also improved significantly. At the 1-year follow-up she was back to her baseline overall condition and showed no recurrence of any visual or neurological symptoms.
Discussion
The neurological manifestations and spinal-cord MRI findings of the patient were consistent with acute myelitis. The differential diagnosis for the etiology of acute myelitis primarily included Behçet disease, sarcoidosis, infections (e.g., syphilis, toxoplasmosis, and viruses), systematic diseases (e.g., lupus erythematosus and Sjogren's syndrome), and neuromyelitis optica. Behçet disease, sarcoidosis, infections, and systematic diseases were excluded by the absence of disease-related clinical manifestations and negative serological tests. neuromyelitis optica was excluded based on previous ophthalmological examinations indicating that the visual impairment was caused by retinal edema and exudates, and the IgG index and VEP being normal.2
To our knowledge there are only two MRI-documented cases of myelitis in VHK disease in the literature,3,4 and our patient (the third case) has the oldest onset age of 50 years (Table 1 and 2). Women were affected in all three cases (one was from Jordan and the other two were from China) with a wide range for the onset age (37, 16, and 50 years, respectively), which is consistent with reports that VKH disease occurs mostly in Native Americans, East Indian, Asian, Middle Eastern, and Hispanic populations, people aged between 20 and 50 years, and women.5,6 Spinal shock was not seen in the three cases, and is possibly less common in myelitis with VKH disease than in idiopathic acute transverse myelitis. Spinal-cord MRI revealed cervical lesions in all three patients, while thoracic changes are more frequent in idiopathic acute transverse myelitis cases. However, too few patients with VKH disease and acute myelitis have been reported to allow firm conclusions to be drawn about potential differences in disease expression. In contrast to our patient, the other two cases did not present with headache and showed normal CSF cells or mild pleocytosis, which may be due to variations in the extraocular appearance, such as meningismus, tinnitus, vitiligo, and alopecia.7 Moreover, the other two patients had already been off steroids before the neurological presentation, which may be explained either as part of the normal course of the disease or adequate corticosteroid therapy not being introduced early enough.7 Although the clinical profile of VKH disease is well-established, little is known about its pathogenesis. Triggering of CD4+ T cells (Th1, T helper 17, and regulatory T cells) reactive to melanocyte-specific proteins [e.g., tyrosinase, tyrosinase-related protein 1, and TRP-2] by an infectious agent is proposed to be involved in the pathogenesis.8 In addition, genetic factors, including HLA-DR4, HLA-DR1, and HLA-DRB1*0405, may also play an important role.8-11 The absence of melanocytes in the spinal cord means that the precise mechanism by which VKH disease leads to acute myelitis is unclear. The effectiveness of steroid therapy in the three cases suggests underlying immunological pathogenic mechanisms, which might involve myelin basic protein.12 The history of upper respiratory tract infection of our patient suggests that infectious factors were involved in the pathogenesis, and the specific geographic distribution of these three cases suggests that genetic background also influences the development of VKH disease with acute myelitis. In summary, new insights into immune responses and genetic abnormalities will help to clarify the pathogenic mechanisms underlying VKH disease with acute myelitis.


 XML Download
XML Download