

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Mutations in the gene encoding phosphoribosyl pyrophosphate synthetase (PRS)-I, PRPS1 (MIM 311850), have been reported in four syndromes: X-linked Charcot-Marie-Tooth disease type 5 (CMTX5, or Rosenberg-Chutorian syndrome; MIM 311070),1,2 PRS-I superactivity (MIM 300661),3 Arts syndrome (MIM 301835),4,5 and nonsyndromic sensorineural deafness (DFN2; MIM 304500).6 PRPS1 belongs to a family that comprises three highly conserved genes: PRPS1, PRPS2 (MIM 311860), and PRPS1L1 (MIM 611566).7,8
The phenotypes associated with PRPS1 mutation cover a wide clinical spectrum.7 The characteristic phenotypes of CMTX5 are peripheral neuropathy, early-onset hearing loss, and optic atrophy.1,2 PRS-I superactivity is characterized by gout with uric-acid overproduction and neurodevelopmental impairment.3 Patients with Arts syndrome present with mental retardation, early-onset hypotonia, ataxia, delayed motor development, hearing impairment, and optic atrophy.4 X-linked postlingual nonsyndromic hearing loss is an isolated symptom in DFN2.6
Charcot-Marie-Tooth disease (CMT), which is also known to be a hereditary motor and sensory neuropathy, is a clinically and genetically heterogeneous disorder of the peripheral nervous system.9 The disease is conventionally classified into demyelinating neuropathy (CMT1), axonal neuropathy (CMT2), and X-linked inheritance (CMTX). CMTX has been further divided into many subtypes (from CMTX1 to CMTX6) according to the clinical symptoms and genetic causes. CMT is reportedly associated with more than 60 causative genes or loci (Inherited Peripheral Neuropathies Mutation Database, http://www.molgen.ua.ac.be/CMTMutations/mutations). However, it is believed that many causative genes remain unidentified. Thus, efficient analytical tools are required to expedite the identification of the underlying genetic causes of this disease.10,11 In the present study, the application of wholeexome sequencing (WES) proved to be an effective strategy for identifying a rare genetic cause in a small pedigree.
The literature contains only one case report of patients with CMTX5.1 The present study applied WES to reveal a novel p.Ala121Gly mutation in PRPS1 in a Korean family with CMTX5 without optic atrophy.
Case Report
The proband (Fig. 1A, IV-1; family ID: FC360) visited our neurology clinic at 15 years of age because of gait disturbance and hearing loss. His parents had first noticed a decreased response to auditory stimulation at an age of 5 months. At 6 years of age he experienced frequent falls and steppage gait caused by weakness of the distal lower extremities. Audiological examinations at 12 years of age revealed bilateral sensorineural hearing loss, and after cochlear surgery he began using a hearing aid. He did not exhibit any symptoms or signs of mental retardation, ataxia, optic atrophy, hypotonia, or hyperuricemia. A neurological examination performed when the patient was 17 years of age revealed weakness and atrophy of the bilateral distal muscles of the lower limbs without proximal muscle involvement. The values on the Medical Research Council scale were G3/5 for the finger abductor, G3/5 for the anterior tibial, and G4/5 for the gastrocnemius muscles. The vibration and position senses were more severely disturbed than the pain and temperature senses. The deep tendon reflexes were absent in all extremities, and no pathological reflexes were observed. Examination of the dilated fundus and visual evoked potentials (VEPs) produced normal findings. The serum level of creatine kinase was elevated (432 IU/L; normal range, 0-185 IU/L), but that of uric acid was within the normal range.
Two male relatives (III-3 and III-5) had CMT phenotypes similar to the proband. A gait problem was noticed in patient III-3 (a 43-year-old man) at the age of 4 years, and this walking difficulty progressed so that he had been confined to a wheelchair since the age of 40 years. His brother (III-5, 39 years of age) experienced similar symptoms at the age of 5 years; however, he was still able to walk with assistance. Both III-3 and III-5 experienced hearing loss before 1 year of age, but dilated fundus and VEP examinations produced normal findings. Neither of these family members agreed to participate in a DNA analysis. Neither the proband's mother (III-2, 45 years of age) nor her relative (II-5, a 68-year-old woman) had hearing loss, gait disturbance, or optic atrophy. Nerve conduction studies of the proband's mother were normal. From history taking it appeared that neither of the deceased relatives of the proband's mother (I-2 and II-3) had complained of gait, hearing, or visual impairments.
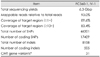
The results of the nerve conduction studies performed in the proband at the ages of 15 and 17 years are given in Table 1. Motor-nerve-conduction studies revealed decreased amplitudes of compound muscle action potentials and slowing of motor conduction velocities in the bilateral median, ulnar, peroneal, and tibial nerves, which were more severe in the lower extremities. Sensory-nerve action potentials could not be elicited in any of the tested nerves, which was compatible with sensorimotor peripheral neuropathy. Needle EMG revealed fibrillation potentials and neurogenic motor-unit action potentials.
The total sequencing yield of WES was approximately 6.3 Gbp, and the coverage rate of the targeted exonic regions (read depth ≥10×) was 83.4%. The number of observed variants was 46301 single-nucleotide polymorphisms (SNPs) and 8158 indels, of which 17409 and 505 were coding SNPs and indels, respectively (Table 2). After functionally significant SNPs in CMT-related genes were selected, the SNPs reported in dbSNP135 (http://www.ncbi.nlm.nih.gov) and 1000 Genomes Project database (http://www.1000genomes.org/) were further filtered out. Subsequent capillary sequencing of the DNA of the proband's mother revealed a novel missense mutation, c.362C>G (Ala121Gly), in exon 3 of PRPS1. The proband and his mother were hemizygous and heterozygous for this mutation, respectively (Fig. 1B), which is compatible with an X-linked recessive inheritance mode. The c.362C>G mutation has not been reported in the worldwide dbSNP135 and 1000 Genomes Project human genome databases, or in the Inherited Peripheral Neuropathies Mutation Database (http://www.molgen.ua.ac.be/CMTMutations/mutations). The mutation was not found in 250 healthy controls, and the site was well conserved among different species (Fig. 1C). Several in silico analyses predicted that the mutation may affect protein function, with scores of 0.01, 0.88, -1.00 in Sorting Intolerant From Tolerant (http://sift.jcvi.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), and MUpro (SVM) program (http://mupro.proteomics.ics.uci.edu/), respectively.
In addition to the PRPS1 c.362C>G mutation, 30 more nonsynonymous variants were identified in CMT-relevant genes (Table 3). However, none of the variants was considered as being causative because they were all also observed in controls or did not fit the X-linked recessive inheritance mode observed within the family. The 17p12 duplication was also excluded by pretesting before exome sequencing.
Discussion
Whole-exome sequencing was used to identify a novel PRPS1 mutation in a family with CMTX with peripheral polyneuropathy and early-onset sensorineural hearing loss. The previously described CMTX5 patients harboring PRPS1 mutations (c.344T>C and c.129A>C) exhibited peripheral neuropathy with segmental demyelination and axonal loss, sensorineural hearing loss from infancy, and visual impairment during the first decade of life, and experienced optic neuropathy at approximately the same age as they developed peripheral neuropathy.1 The clinical phenotypes of the family presented herein are similar to those reported CMTX5 patients; 1,2 however, the present patients did not have optic atrophy. These findings suggest that the severity and progression of CMTX5 phenotypes vary according to the sites of the PRPS1 mutations.
Defects in X-linked PRPS1 manifest as PRS-I enzyme overexpression (PRS-I superactivity), severe PRS-I deficiency (Arts syndrome), moderate PRS-I deficiency (CMTX5), and mild PRS-I deficiency (DFN2).1-6 PRS-I superactivity is an X-linked disease associated with hyperuricemia and hyperuricosuria.3 Neurodevelopmental impairments, particularly sensorineural deafness, have been reported occasionally.12 Arts syndrome has the most disabling clinical manifestations of PRPS1 missense mutations, resulting in severe functional loss of the protein.5 These patients harbor neurological deficits, including mental retardation, ataxia, hypotonia, delayed motor development, optic atrophy, peripheral neuropathy, and sensorineural deafness, which result in early death (during the first decade of life).4 Moderately decreased PRS-I activity caused by a PRPS1 missense mutation is known as CMTX5 or Rosenberg-Chutorian syndrome.1,2 Early sensorineural deafness, peripheral neuropathy, and optic neuropathy together constitute a typical symptom triad of this disease, but variations in disease duration and severity have been reported.13 DFN2 caused by PRPS1 mutations manifests as progressive nonsyndromic hearing loss.6 The patient described herein displayed peripheral sensorimotor neuropathy and early-onset sensorineural deafness, but he did not exhibit mental retardation, optic atrophy, hypotonia, or hyperuricemia. In addition, his middle-aged male relatives with the same manifestations did not complain of any visual disturbances. Therefore, his symptoms did not correspond accurately with any of the four known distinct syndromes associated with PRPS1 mutation, strongly suggesting that the clinical spectrum of diseases with PRPS1 mutation is wider than previously described.
PRS, which is an enzyme that is distributed in all human tissues, occurs in two isoforms (PRS-I and II), which are encoded by the highly conserved genes PRPS1 and PRPS2, respectively. 14 PRS-I catalyzes the synthesis of phosphoribosyl pyrophosphate, which is an essential cofactor for both the synthesis and salvage of purine, pyrimidine, and pyridine nucleotides.14 Thus, PRPS1 mutation affects vital cell functions, including nucleic acid synthesis, energy metabolism, and cellular signaling.7 Based on the primary sequence alignment with PRS-1 from other species and on its quaternary structure, the location of Ala121 does not seem to be an allosteric regulatory site. Rather, it is located in the vicinity of the intracellular interacting region and catalytic site. Thus, reduced enzymatic activity might be derived from a loose interaction between the hexamers or the slightly altered catalytic site associated with Ala121 mutation.
The results of several recent studies suggest that mutant PRPS1-mediated disease can be treated by improving nucleotide metabolism; supplementation of the diet with S-adenosyl methionine (SAM) in patients with Arts syndrome yielded improvements in their condition.7 Nucleotides such as purines are usually not absorbed in the body because of oxidation to uric acid; however, pyrimidines can be absorbed by oral intake through the intestinal tract. Nevertheless, unlike most purines, SAM can cross both the gut and the blood-brain barrier, and has been successfully used in the treatment of depression, dementia, and osteoarthritis.15 The improvement in the symptoms of Arts syndrome after SAM supplementation implies that this treatment may also alleviate some of the symptoms of patients with other PRPS1 mutations by replenishing purine nucleotides.7 These clinical data imply the applicability of this therapeutic approach in the patients described herein.
In summary, a novel PRPS1 mutation was identified in a family with CMTX5 using WES. The findings of this study will expand the clinical spectrum of diseases with PRPS1 mutation and will be useful for the molecular diagnosis of clinically heterogeneous peripheral neuropathies.



 XML Download
XML Download