

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
GNE myopathy, otherwise known as Nonaka myopathy or hereditary inclusion-body myopathy, is an autosomal recessive myopathy characterized by weakness of the distal muscles in the lower limbs with preferential involvement of the tibialis but a sparing of quadriceps muscles, with early-adult onset at ages ranging from 15-40 years. The serum creatine kinase (CK) level is normal or mildly elevated, and muscle biopsy typically shows rimmed vacuoles (RV).1
GNE myopathy is caused by mutations in the gene encoding UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE). This gene encodes a bifunctional protein with two enzymatic activities: UDP-GlcNAc2-epimerase and ManNAc kinase (MNK). Although the molecular mechanism by which the mutations in the GNE gene cause the muscle degeneration seen in GNE myopathy remains unclear, it has been proposed that the mutations lead to defective sialylation of muscle. It has also been reported that there is an increased requirement for sialic acid during pregnancy in patients with GNE myopathy.
We describe a GNE myopathy patient carrying a GNE mutation (D208N/M29T) who presented a rapid deterioration in strength of the distal lower limbs during pregnancy.
Case Report
A 27-year-old Korean woman complained of difficulty walking during her first pregnancy. She found it more comfortable to walk on her toes than on her heels. She subsequently experienced impaired foot dorsiflexion with frequent falling and rapid wasting of both anterior tibialis muscles during the third trimester and following delivery. A review of her history revealed normal mental and physical development, and she reported no difficulties with daily activities prior to pregnancy. Her family history was significant in having an older sister with a similar progressive limb weakness, appearing in the lower limbs at 19 years and in the upper limbs at. The neurologic examination of our case revealed marked weakness and atrophy of both anterior tibialis muscles with no weakness of the quadriceps and upper limbs. Motor strengths were decreased in ankle dorsiflexion [1/5 on the Medical Research Council (MRC) Scale] and plantar flexion (4/5 on the MRC Scale). She had normal deep tendon reflexes in all limbs without sensory deficit. She had no pathologic reflexes. The serum CK level was slightly elevated to 302 U/L (normal range 21-215 U/L). Electromyography revealed myopathic changes.
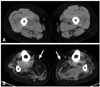
A computed tomography scan of her skeletal muscles showed severe fatty infiltration of the gastrocnemius, soleus, and tibialis anterior muscles with sparing of the quadriceps muscles (Fig. 1). A muscle biopsy performed on the vastus lateralis muscle revealed mild myopathic changes without RV. After obtaining informed consent, 11 coding exons (exons 2-12) of the GNE gene were analyzed, which revealed compound heterozygous mutations changing the ATG (methionine) to ACG (threonine) at codon 29 (M29T) in exon 2 and the GAT (aspartic acid) to AAT (asparagine) at codon 208 (D208N) in exon 4 (Fig. 2).
Discussion
UDP-GlcNAc2-epimerase catalyzes the rate-limiting step in sialic acid biosynthesis and MNK catalyzes the subsequent step. Sialic acids, which are N-acetylated derivatives of neuraminic acid, are the most abundant terminal monosaccharides on the glycoconjugates of eukaryotic cell surfaces, and they play important roles in development, regeneration, and biomedical functioning.2,3
The main pathophysiology involved in GNE myopathy is hyposialylation and abnormal glycosylation, which may lead to the misfolding of some glycoproteins. Noguchi et al.4 demonstrated that the levels of sialic acid in muscles and primary cultured cells from GNE myopathy patients were reduced by 60-75% compared to healthy controls. Correction of this hyposialylation has been attempted in GNE myopathy mice and could led to a therapeutic strategy for human patients.5 The sialylation status in skeletal muscle tissue is also greatly altered, especially in the fibers with RV, suggesting a close relationship between hyposialylation and the formation of RV.4,6
We have reported a Korean patient with GNE myopathy who carried the compound heterozygous mutation of the epimerase domain of the GNE gene. Although the M29T mutation has been reported previously,7 the D208N mutation found in the present case is novel. Various GNE mutations have been identified in GNE myopathy patients from various ethnic groups. The M712T mutation is the most common mutation in Jewish hereditary inclusion-body myopathy, while the most frequent mutation in Japanese and Korean GNE myopathy patients is the V572L mutation, which suggests that a common founder effect exists in these populations.
It was especially interesting that our patient experienced rapid deterioration in muscle strength during her first pregnancy. Similar cases have been reported previously.8,9 Sialic acid is known to be essential for embryonic development.10 and it has been suggested that the elevated sialic acid level in pregnant women and the high level of GNE expression in the placenta may indicate an increased requirement for sialic acid during pregnancy. The lower levels of sialic acid in women with GNE myopathy compared to healthy controls could explain the clinical deterioration during pregnancy observed in our patient. We therefore suggest that the rapid progression of muscle weakness in our patient during pregnancy may be explained by hyposialylation11 and abnormal glycosylation associated with GNE myopathy and an increased requirement for sialic acid during pregnancy.
We have reported a representative case with rapid deterioration in muscle strength during pregnancy in GNE myopathy. However, future studies should involve more patients and address causality in order to allow definitive conclusions to be drawn.

 XML Download
XML Download