

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Migraine is a heterogeneous neurological disorder characterized as a recurring, episodic, unilateral headache. Migraine pain usually throbs1 and typically intensifies during physical activities that increase intracranial pressure (e.g. bending over; coughing).2,3 The pain is associated with high incidence of nausea and predominance of hypersensitivity to light (photophobia) and noise (phonophobia). Symptoms of lesser prevalence include, aversion to odors (osmophobia), vomiting, fatigue, red eyes, tearing, nasal congestion, frequent yawning. A migraine attack may be precipitated by endogenous factors (i.e., hormonal changes, psychosocial stress, sleep deficit or surplus, hunger), or by exogenous factors (i.e., certain kinds of food; stimulation of different sensory modalities). An attack can be preceded by abnormal visual, sensory, motor and/or speech functions (migraine with aura) or start with no warning signs (migraine without aura).
Vascular Theory of Migraine-Extracranial Origin
For many years, migraine headache has been thought to be related to dilation of extracranial arteries. The theory was, as cited extensively in medical textbooks, that abnormal vasodilatation during migraine causes mechanical activation of perivascular stretch receptors, resulting in throbbing headache.4 This view was based on observations that extracranial arteries are vasodilated, edematous, and partially damaged during migraine.4,5 However, the extracranial vascular theory has fallen out of favor because clinical studied have yielded no convincing evidence for any significant extracranial vasodilation during migraine, nor have they shown that vasodilation can produce headache.6,7
Vascular Theory of Migraine-Intracranial Origin
The prevailing view today is that migraine headache is a neurovascular disorder of intracranial origin that involves meningeal blood vessels and the pain fibers that innervate them. This theory has originated from reports that electrical and mechanical stimulation of dural vasculature (but not the surface of the brain) produced referred head pain in awake patients undergoing craniotomy8,9: 1) periorbital pain - by stimulating the superior sagittal sinus or blood vessels of the floor of the anterior fossa; 2) parietal/temporal pain - by stimulating the middle meningeal artery; 3) occipital pain - by stimulating the dura at the floor of the posterior (PO) fossa, and the sigmoid, transverse and occipital sinuses. It should be emphasized that a) no sensation other than pain was evoked by stimulation of these structures; b) stimulation of non-vascular areas of the dura was largely ineffective in inducing pain sensation. These findings fit well with the pattern of dural innervation, whereby sensory nerves that originate in trigeminal and upper cervical ganglia closely follow meningeal blood vessels but not non-vascular areas of the dura.9 It was not until the 1980's that the nature of dural innervation was proved to be nociceptive. We now know that the dura is richly innervated by unmyelinated (C-fibers) and thinly myelinated (Aδ fibers) axons that originate in the trigeminal ganglion and in C1-3 dorsal root ganglions6,10-12 and that these pain fibers contain vasoactive neuropeptides such as substance P and calcitonin gene-related peptide (CGRP).13,14 These lines of evidence promoted the theory that the headache phase of migraine is mediated by activation of nociceptors that innervate meningeal blood vessels (i.e., meningeal nociceptors), and provided the basis for developing animal models of neurovascular head pain with intracranial origin.
Experimental Activation of Trigeminovascular Pathways
The first animal model of neurovascular head pain employed the paradigm of electrical and/or mechanical stimulation of the dural sinuses.15,16 Using anatomical, physiological, histological and pharmaceutical techniques, such animal studies have demonstrated the presence of dura-sensitive neurons in brain and spinal cord structures, such as the medullary dorsal horn, thalamus, hypothalamus, periaqueductal gray (PAG).15,17-24 In the medullary dorsal horn25,26 and thalamus27,28 the majority of these trigeminovascular neurons respond to noxious skin stimuli either preferentially (wide-dynamic range neurons) or exclusively (high-threshold neurons). Administration of anti-migraine drugs such as ergotamines, triptans, or CGRP antagonists have been shown to inhibit responses to electrical stimulation of the dura in dura-sensitive neurons in the medullary dorsal horn.19,29-31
The acute stimulation of dural sinuses as a model for neurovascular head pain with intracranial origin has identified the trigeminovascular system and established the potential involvement of each of its elements in vascular headache and the associated symptoms. However, acute electrical or mechanical stimulation of the dura does not induce migraine in man, and can only evoke a short burst of activity in peripheral and central trigeminovascular neurons.
In recent years, it has become apparent that many types of prolonged or chronic pain are associated with long-lasting activation and sensitization of peripheral nociceptors and/or central nociceptive neurons in the dorsal horn. Incorporating these concepts into basic research on migraine pathophysiology, a new animal model for long-lasting headache of migraine was developed.
This model involves prolonged activation and subsequent sensitization of the trigeminovascular system in response to a brief exposure of the dura to a mixture of inflammatory agents consisting of serotonin, bradykinin, histamine, and prostaglandin.32 These agents activate and sensitize somatic and visceral nociceptors in the rat33-37 and are potent algesics in humans,38-41 capable of inducing headache.41
Peripheral Sensitization
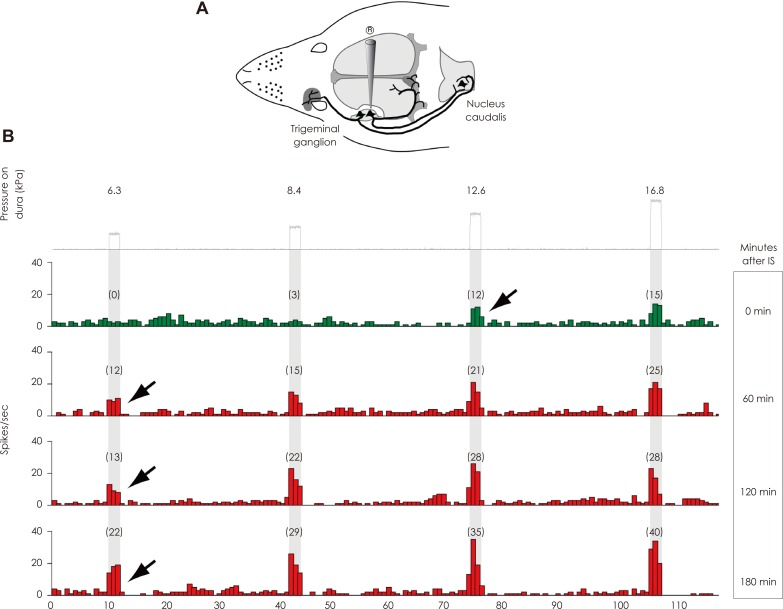
Using this animal model, it was found that a brief chemical irritation of the dura activates and sensitizes meningeal nociceptors (first-order trigeminovascular neurons) over a long period of time, rendering them responsive to mechanical stimuli to which they showed only minimal or no response prior to their sensitization (Fig. 1).32 During migraine, such peripheral sensitization is likely to mediate the throbbing pain and its aggravation during routine physical activities such as coughing, sneezing, bending over, rapid head shake, holding one's breath, climbing up the stairs, or walking.
By the end of migraine, when meningeal nociceptors are presumably no longer sensitized, their sensitivity to fluctuations in intracranial pressure returns to normal, and the patient no longer feels throbbing.
Central Sensitization
Medullary dorsal horn
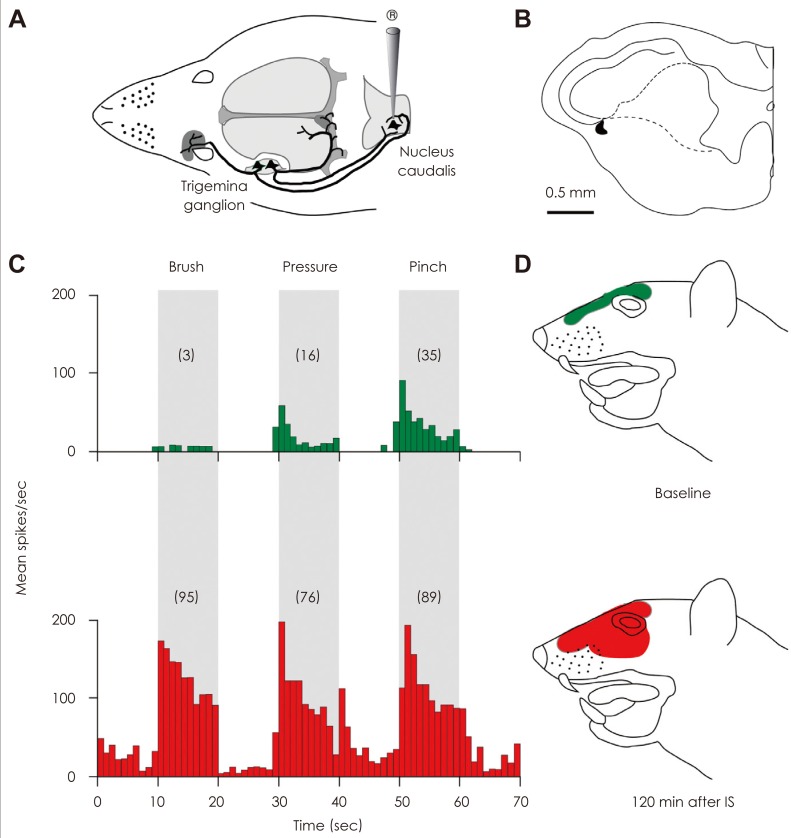
Brief stimulation of the dura with inflammatory agents also activates and sensitizes second-order trigeminovascular neurons located in the medullary dorsal horn that receive convergent input from the dura and the skin.25 In this paradigm, the central trigeminovascular neurons develop hypersensitivity in the periorbital skin, manifested as increased responsiveness to mild stimuli (brush, heat, cold) to which they showed only minimal or no response prior to their sensitization (Fig. 2). The induction of central sensitization by intracranial stimulation of the dura, and the ensuing extracranial hypersensitivity was taken to suggest that a similar process occurs in patients during migraine (Fig. 3).
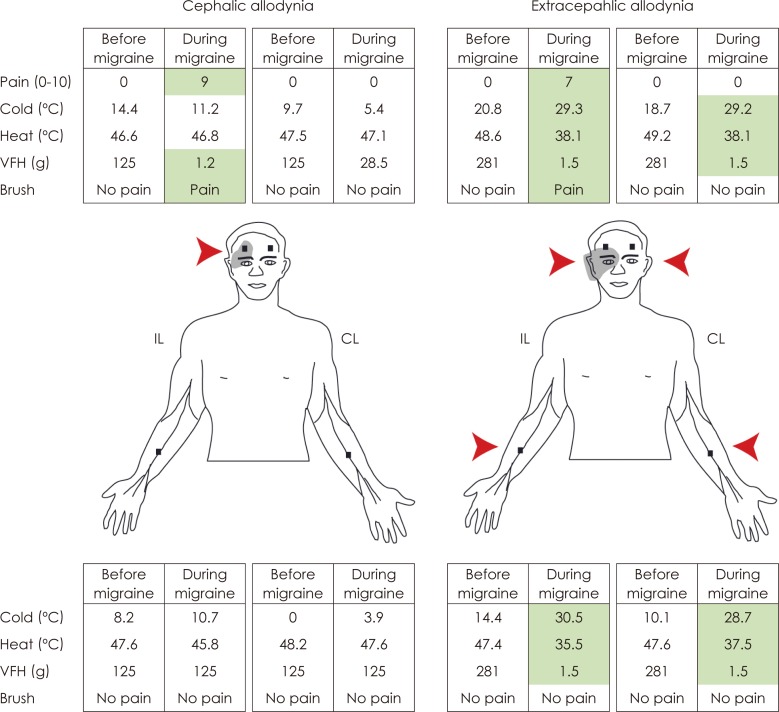
Extracranial hypersensitivity during migraine was first noted in 187342 and later documented in the 1950's.5,43 At that time, extracranial hypersensitivity was ascribed to "hematomas that develop hours after onset of headache as a result of damage to vascular walls of blood vessels such as the temporal artery",5 or "widespread distension of extracranial blood vessels or spasm of suboccipital scalp muscles".43 The current view, however, is that extracranial hypersensitivity is a manifestation of central neuronal sensitization rather than extracranial vascular pathophysiology. Recent quantitative stimulation applied to the surface of the skin showed that pain thresholds to mechanical, heat, and cold skin stimuli decrease significantly during migraine in the majority of patients (Fig. 3A).44 This skin hypersensitivity, termed cutaneous allodynia, is typically found in the periorbital area on the side of the migraine headache. Patients commonly notice cutaneous allodynia during migraine when they become irritated by innocuous activities such as combing, shaving, taking a shower, wearing eyeglasses or earrings, or resting their head on the pillow on the headache side. Ipsilateral cephalic allodynia is likely to be mediated by sensitization of trigeminovascular neurons in the medullary dorsal horn that process sensory inputs from the dura and periorbital skin.
Thalamus
In the course of studying cephalic allodynia during migraine, we unexpectedly found clear evidence for allodynia in remote skin areas outside the innervation territory of the trigeminal nerve (Fig. 3B).44
In the discussion of that study, we proposed that ipsilateral cephalic allodynia is mediated by sensitization of dura-sensitive neurons in the medullary dorsal horn because their cutaneous receptive field is confined to innervation territory of the ipsilateral trigeminal nerve21,25,26,45-47 and that extracephalic allodynia must be mediated by neurons that process sensory information they receive from all levels of the spinal and medullary dorsal horn. Our search of such neurons focused on the thalamus since an extensive axonal mapping of sensitized trigeminovascular neurons in the spinal trigeminal nucleus revealed distinguish projections to the PO, the ventral posteromedial and the sub-parafascicular nuclei.
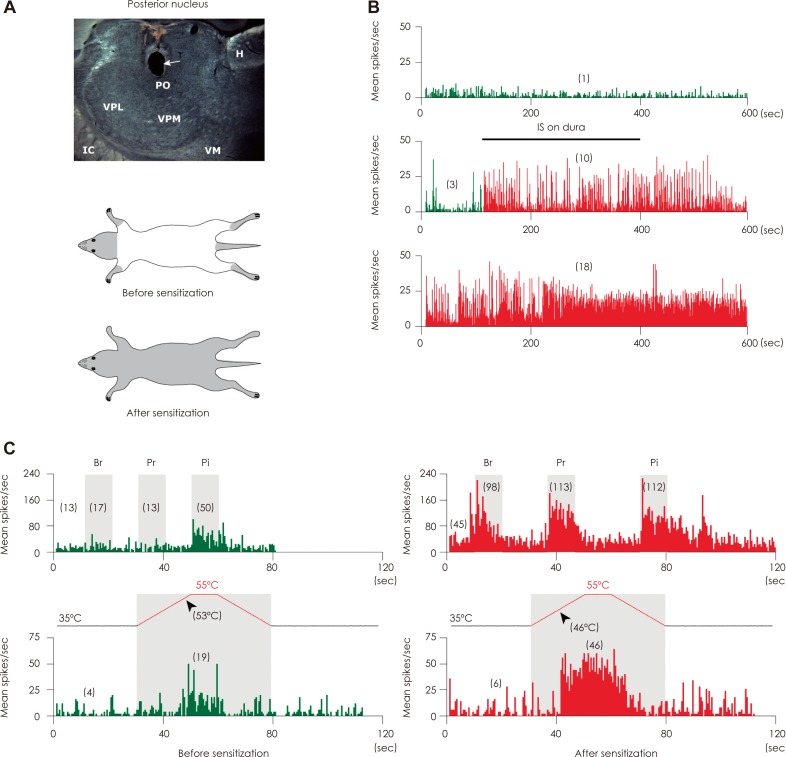
In 2010 we reported that topical administration of inflammatory molecules to the dura sensitized thalamic trigeminovascular neurons that process sensory information from the cranial meninges and cephalic and extracephalic skin.48 Sensitized thalamic neurons developed ongoing firing and exhibited hyper-responsiveness (increased response magnitude) and hypersensitivity (lower response threshold) to mechanical and thermal stimulation of extracephalic skin areas (Fig. 4).
Relevant to migraine pathophysiology was the finding that in such neurons, innocuous extracephalic skin stimuli that did not induce neuronal firing before sensitization (e.g., brush) became as effective as noxious stimuli (e.g., pinch) in triggering large bouts of activity after sensitization was established.
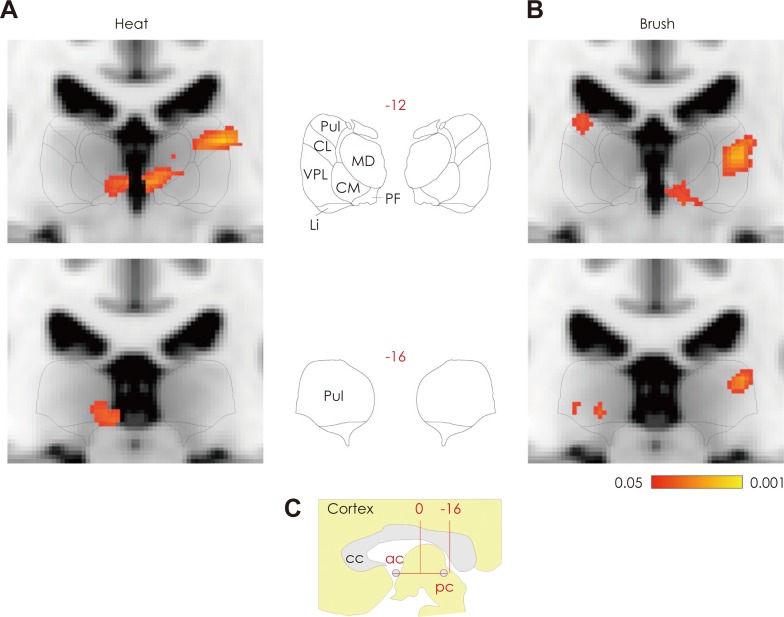
To understand better the transformation of migraine headache into widespread, cephalic and extracephalic allodynia, we also studied the effects of extracephalic brush and heat stimuli on thalamic activation registered by fMRI during migraine in patients with whole-body allodynia.48 Functional assessment of blood oxygenation level-dependent signals showed that brush and heat stimulation at the skin of the dorsum of the hand produced larger blood oxygenation level-dependent responses in the PO thalamus of subjects undergoing a migraine attack with extracephalic allodynia than the corresponding responses registered when the same patients were free of migraine and allodynia (Fig. 5).
Temporal aspects of sensitization and their implications to triptan therapy
Central sensitization can be either activity-dependent or activity-independent.49 The induction of sensitization in second-order trigeminovascular neurons, using chemical stimulation of the rat dura, is activity dependent, as evidenced by lidocaine blockade of afferent inputs from the dura. Once established, however, sensitization of the second-order trigeminovascular neurons becomes activity-independent, as it can no longer be interrupted by lidocaine on the dura.25 Translating theses findings in the context of migraine with allodynia, it appears that central sensitization depends on incoming impulses from the meninges in the early phase of the attack, and maintains itself in the absence of such sensory input later on. This view is strongly supported by the effects of the anti-migraine 5-HT1B/1D agonists, known as triptans, on the induction and maintenance of central sensitization in the rat,50 and the corresponding effects of early and late triptan therapy on allodynia during migraine.51 In the rat, triptan administration concomitant with chemical irritation of the dura effectively prevents the development of central sensitization. Similarly, treating patients with triptans early, within 60 min of the onset of migraine, effectively blocks the development of cutaneous allodynia. However, neither central neuronal sensitization in the rat, nor cutaneous allodynia in patients, can be reversed by late triptan treatment (2 hours after the application of sensitizing agent to the dura in the animal model, and 4 hours after the onset of migraine in allodynic patients). Most importantly, central sensitization appears to play a critical role in the management of migraine headache of allodynic patients. While non-allodynic patients can be rendered pain-free with triptans at any time during an attack, allodynic patients can be rendered pain-free only if treated with triptans early in the attack, namely, before the establishment of cutaneous allodynia.51
The findings that triptans cannot block ongoing sensitization in second-order trigeminovascular neurons is consistent with the evidence that these neurons do not posses the 5-HT1D receptor52 that mediates the neuronal action of these drugs. If triptans do not abort migraine in the presence of allodynia (i.e., central sensitization), how do they render the patient pain-free in the absence of allodynia? The simple option would be a peripheral action of triptans that suppresses of ongoing sensitization in the meningeal nociceptors that bombard the second-order neuron in the dorsal horn. However, the evidence shows that sensitized meningeal nociceptors are not inhibited by triptans.53 Thus, it appears that triptans abort migraine by a central, presynaptic action in the dorsal horn that blocks transmission of nociceptive signals between first- and second-order trigeminovascular neurons.
Modulation of Central Sensitization
A growing body of evidence suggests that migraine patients are mostly non-allodynic during the first years of their migraine experience, and are eventually destined to develop allodynia during their migraine attacks.44,51,54 It is therefore possible that repeated migraine attacks over the years have cumulative adverse consequences on the function of the trigeminovascular pathway, one of which is susceptibility to develop central sensitization. The threshold for a central trigeminovascular neuron to enter a state of sensitization depends on the balance between incoming nociceptive signals and their modulation by spinal and supraspinal pathways. Many of the modulatory supraspinal pathways converge on the PAG and rostral ventromedial medulla (RVM).55 Recent imaging studies have shown that the PAG is activated during migraine56 and that it is deposited with abnormally high levels of iron in patients with a long history of migraine, suggesting an abnormal neuronal functioning.57 Abnormal PAG functioning can either enhance activity of RVM neurons that facilitate pain transmission in the dorsal horn, or suppress activity of RVM neurons that inhibit pain transmission in the dorsal horn.58 This may enhance excitability and, therefore, promote responses of second-order trigeminovascular neurons to incoming nociceptive signals from the meninges, resulting in a reduced threshold for entering a state of central sensitization. Furthermore, the transition from episodic to chronic migraine that occurs in some patients over the years may involve a shift in the underlying pathophysiology from transient to chronic state of sensitization. Altered functions of modulatory supraspinal pain pathways can contribute to this progression in migraine pathophysiology.
Brainstem Generator Theory
A rather radical view on migraine pathophysiology originated from a 1987 report suggesting that the PAG may constitutes a so-called "headache generator". In that study, 15 out of 175 pain patients (8.5%) developed migraine-like headache immediately after undergoing craniotomy for electrodes implantation at (or near) the PAG.59 This point of view has promoted an interpretation that activation of the PAG during migraine is the source, rather than the consequence, of migraine pain.56,60 However, numerous studies have yielded overwhelming evidence to disprove the concept of the PAG as a "headache generator". The first line of evidence is that persistent postoperative headache lasting 3 months or longer is routinely observed in 9-38% of patients undergoing craniotomy for a wide variety of procedures, with or without electrode placement.61-65 This crucial information was not available to Raskin et al.59 when they made the statement that "comparable headache syndromes have not been seen following craniotomy or burr hole placement performed for a variety of disorders."
The second line of evidence comes from neuroimaging studies in humans66-69 and Fos expression studies in animals,70 demonstrating that PAG activation occurs in numerous non-headache pain paradigms. This indicates that PAG activation is a universal consequence of nociceptor activation anywhere in the body, which is perfectly consistent with the multiple input arriving at the PAG from nociceptive neurons through the length of the spinal cord. The third line of evidence is that electrical stimulation of the PAG produces a general whole-body pain relief.71 This indicates that modulation of pain by the PAG is non-specific, which is consistent with the anatomical evidence that individual RVM neurons project to multiple segments of the spinal cord and terminate in the dorsal horn.72-75 Finally, the fourth line of evidence is that stimulation of the PAG or RVM cannot generate firing in spinothalamic tract neurons; it can only increase or decrease firing in responses to noxious stimulation of their peripheral receptive fields.58 Accordingly, it is activation of specific dorsal horn neurons by input they receive from peripheral nociceptors that determine where pain modulation is needed. In the case of migraine it is activation of trigeminovascular neurons in the medullary dorsal horn by inputs from meningeal nociceptors.
Activation of Meningeal Nociceptors
In spite of extensive research, the endogenous mechanisms by which meningeal nociceptors become activated have not yet been identified. We shall briefly review two hypotheses that attempt to describe endogenous cascades of events in the dura that may lead up to activation of meningeal nociceptors during the headache phase of migraine.
Neurogenic Inflammation Hypothesis
According to this hypothesis, meningeal nociceptors are activated by local release of endogenous inflammatory mediators through a process termed neurogenic inflammation.6,76 The process refers to a host of events, including local increase in blood flow, leakage of plasma protein from blood vessels, mast cell degranulation, and platelet aggregation. Neurogenic inflammation can be evoked experimentally by noxious sti-mulation of nociceptors that innervate a given tissue and the subsequent release of vasoactive neuropeptides such as substance P, neurokinin A, and CGRP.77 Similar markers of neurogenic inflammation and neuropeptide release have been induced in the rat dura by antidromic activation of meningeal afferents through electrical stimulation of the trigeminal ganglion.78,79 Support for the role of neurogenic inflammation in migraine comes from evidence that plasma protein extravasation and mast cell degranulation can be blocked in the rat by antimigraine drugs, such ergot alkaloids and triptans,78,80 and by non-steroidal anti-inflammatory agents, such as indomethacin and acetylsalicylic acid.81
The neurogenic inflammation hypothesis does not explain, however, what causes the initial activation of meningeal nociceptors that triggers neurogenic inflammation in the dura. The next hypothesis has been proposed in an attempt to address this dilemma.
Cortical Spreading Depression Hypothesis
Cortical spreading depression (CSD) refers to a wave of brief excitation followed by prolonged (15-30 min) inhibition of neuronal activity that propagates slowly across the cortex at a rate of 2-6 mm/min. This neural phenomenon was first observed by Leao in the cortex of anesthetized rabbits,82 and later correlated with localized changes in blood flow that spread through the cortex at a similar rate.83 CSD has been implicated in the pathophysiology of migraine on the basis of neuroimaging studies showing slowly-migrating changes in cortical blood flow in patients tested during the visual aura phase of migraine.83-86 Since aura precedes the onset of headache by 20-30 min, it was postulated that CSD may lead up to the initial activation of meningeal nociceptors. But how would abnormal cortical activity produce an impact on dural pain fibers and blood vessels across the pia mater and arachnoids?
Direct electrophysiological evidence for the activation of trigeminovascular neurons by CSD were reported recently,87,88 the CSD hypothesis relies on two suppositions that are based on two independent sets of observation. One assumption is that molecules such as potassium ions, hydrogen ions, and glutamate that are released extracellularly during CSD in the cortex86,89,90 diffuse through the overlying meninges and activate meningeal nociceptors. The other assumption is that the induction of neurogenic inflammation in the dura by CSD91 is mediated by antidromic axonal reflex that propagates through sensory trigeminal axons collateral that innervate both the pia and dura. This assumption may be consistent with the finding that sensory denervation of the meninges blocks CSD-induced neurogenic inflammation in the dura.91 Anatomical evidence for the existence of individual trigeminal axons that innervate both the pia and dura are now available.92
Concluding Remarks: Implications for Migraine Therapy
Based on current understanding of migraine-associated cutaneous allodynia and the most probable mechanism of action of triptans in migraine therapy, it is recommended that patients with allodynia take triptans as early as possible into the attack before the emergence of any sign of allodynia. This recommendation is consistent with patients' testimonies that triptans are much more likely to render them pain-free when taken early rather than late.
Nevertheless, most migraineures testify that they routinely delay treatment until attacks are fully developed or the pain is severe. Justifying the delayed treatment are concerns about side effects, addiction, limits on supply imposed by prescribers, cost, and most commonly waiting to see if headache develops into a severe migraine attack.93 For these patients, one way to terminate migraine with allodynia and fully developed central sensitization is parenteral administration of COX1/COX2 inhibitors.94 Infusion of the COX1/COX2 inhibitor ketorolac in allodynic patients who already missed the critical period for triptan therapy terminated both the headache and the allodynia provided that the patient had no history of using opioids to treat her/his migraines. In the rat, infusion of COX-1/COX-2 inhibitors blocked sensitization in meningeal nociceptors and suppressed ongoing sensitization in spinal trigeminovascular neurons, suggesting that parenteral nonsteroidal anti-inflammatory drug administration acts in the dorsal horn to inhibit the central neurons directly and reduce the synaptic input from the peripheral trigeminovascular neuron.94,95
Though impractical as a routine migraine therapy, parenteral nonsteroidal anti-inflammatory drug administration should be useful as a non-narcotic rescue therapy for migraine in the setting of the emergency department. Patients who use an opioid therapy over extended period of time are at high risk of developing medication-overuse headache and low response to non-narcotic drugs. The rational for recommending against the use of opioids in allodynic migraine patients is based on evidence that opioids can facilitate sensitization in the dorsal horn96-99 through: 1) upregulation of NMDA receptors function, 2) downregulation of glutamate transporters, 3) production of nitric oxide, 4) activation of spinal glia, and 5) increase extracellular level of prostaglandins.
 XML Download
XML Download