

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Leber's hereditary optic neuropathy (LHON) is characterized as a maternally inherited loss of central vision with optic atrophy due to mitochondrial DNA (mtDNA) mutations.1,2 LHON may be accompanied by other complicating neurological disorders, such as dystonia, parkinsonism, cerebellar ataxia, and myoclonus, and is usually referred to as "LHON-plus syndrome".3-6
Point mutations in genes that encode the complex I subunit of mtDNA are thought to be a cause of this disease, and G11 778A in the NADH dehydrogenase (ND) subunit 4 (ND4) gene (ND4) is the most common mutation.7-9 Although many other types of mutations (including T14484C and G3460A) have also been reported as the cause of LHON,7 the association between the clinical features and these mutations in the mtDNA is unclear. We report herein a case of LHON with olivocerebellar degeneration due to a heteroplasmic G11778A mutation in mtDNA ND4 and a homoplasmic T3394C mutation in the mtDNA ND1 gene (ND1).
Case Report
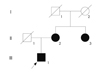
The proband (III-1) (Fig. 1) was a 37-year-old man with severe dizziness and double vision. He had first experienced visual and gait disturbances at 10 years of age. The neurological examination performed on admission revealed mild disturbance of cognitive function (Revised Wechsler Adult Intelligence Scale: total IQ=73, performance IQ=58, verbal IQ=91). Neurological disturbances were observed including bilateral exotropia, double vision, incomplete horizontal movement of the eyes to the bilateral side, horizontal, and vertical gaze-evoked nystagmus, and dysarthria. The light reflex was prompt. No disturbances in cranial nerves I, VII, VIII, and XII were detected. Tremor appeared in his neck, but other involuntary movements including palatal myoclonus were not observed. While his upper and lower limbs showed no paralysis, they exhibited severe cerebellar ataxia and hypotonia. No abnormal findings were detected in his deep tendon reflex and sensory system. Ophthalmological examination revealed atrophy of the optic nerve, but there were no pigmentation changes of the retina. Blood and cerebrospinal fluid analyses were normal. Ergometer exercise did not up-regulate his serum lactate and pyruvate.
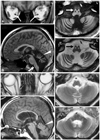
Orbital MRI revealed atrophy of the optic nerve (Fig. 2A), and brain MRI disclosed severe atrophy of the cerebellum and mild atrophy of the brain stem (Fig. 2B). The bilateral inferior olivary nucleus exhibited low signal intensities on T1-weighted imaging, and high signal intensities on T2-weighted imaging, suggesting degeneration (Fig. 2C and D). The patient was diagnosed as having LHON plus olivocerebellar degeneration. Although the thyrotropin-releasing drug taltirelin did not relieve his symptoms, adenosine triphosphate disodium reduced his dizziness.
The patient's mother (II-2) and uncle (II-3) also had optic neuropathy, but other neurological abnormalities such as ataxia and dystonia were not observed. The patient's mother has a history of subarachnoid hemorrhage. MRI of his mother disclosed mild atrophy of the optic nerve (Fig. 2E), pons, and cerebellum (Fig. 2F-H). No signal changes were observed in the inferior olivary nucleus (Fig. 2F-H).
We were unable to confirm the detailed clinical information of the proband's grandmother (I-2).
Mutation analyses of mtDNA
Blood samples were obtained from the patient and his mother with their informed consent, and the methods used were approved by the institutional review board of Tottori University Hospital. Both mtDNA and genomic DNA were extracted by standard procedures. The polymerase chain reaction (PCR) was carried out using the primers 5'-CCTCCCTACTATGCCTAGAAGGA-3' and 5'-TTTGGGTTGTGGCTCAGTGT-3' for ND4, including 11778G analysis, and 5'-AGTTCAGACCGGAGTAATCCAG-3' and 5'-AGGGTTGTAGTAGCCCGTAG-3' for ND1. The primer set for ND4 was designed to identify G11778A mutations, which is the main mutation for LHON. The primer set for ND1 was designed to detect not only the T3394C mutation as a minor mutation for LHON but also an A3243G mutation that is frequently detected in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. PCR products that included the previously reported candidate abnormal points were analyzed by capillary electrophoresis using an automated DNA sequencer. The G11778A and T3394C mutations were identified, while the A3243G mutation was not detected.
The mutations in the mtDNA were confirmed by performing PCR-restriction fragment length polymorphism (RFLP), in which the PCR products were digested using either HaeIII (for T3394C) or Tsp45I (for G11778A).
In order to quantify the heteroplasmic mutation of G117 78A, we prepared vector constructs including 11778G or 11 778A, and semiquantitative analyses of G11778A were performed using a mixture of each with several rate standards (described in Fig. 4).
Sequence analysis revealed the homoplasmic T3394C mutation of the mtDNA (Fig. 3A). This mutation causes a Tyr-to-His amino acid substitution in ND1. PCR-RFLP data revealed that the T3394C mutation present in both the proband (III-1) and his mother (II-2) was homoplasmic; differences between the patient and his mother were not observed for this mutation (Fig. 3B).
The G11778A mutation, which causes an Arg-to-His amino acid substitution in ND4, was also observed (Fig. 4A). PCR-RFLP data showed that this mutation in the patient and his mother was heteroplasmic (Fig. 4B). The semiquantitative analysis performed to determine the effect of this mutation on disease severity revealed that III-1 had a 92% heteroplasmic G11778A mutation, and II-2 had a 70% heteroplasmic G11778A mutation (Fig. 4C).
Established genetic abnormalities associated with cerebellar ataxia including polyglutamine diseases were not found.
Discussion
While visual disturbance is the only symptom observed in most patients with LHON, some cases with other neurological abnormalities have been reported as LHON-plus syndrome. These cases include optic neuropathy with dystonia, parkinsonism, cerebellar ataxia, and myoclonus.3-6 The present case exhibited optic neuropathy with horizontal gaze palsy, gaze-evoked nystagmus, and cerebellar ataxia, and olivocerebellar degeneration on neuroimaging. Genetic analyses identified a heteroplasmic G11778A mutation in ND4 and a homoplasmic T3394C mutation in ND1 of mtDNA.
The most common mutation of LHON is G11778A in the mtDNA, which causes an Arg-to-His amino acid substitution in ND4. Several other mutations with LHON have also been reported. Mutations in ND4, a subunit of mitochondrial complex I, causes complex I deficiency, overproduction of reactive oxygen species, and bioenergetic impairment.10-12 However, the clinical features associated with the G11778A muta-tion and several other mutations in the mtDNA vary,7,13,14 and so the clinical phenotype may be complicated if other mutations are present in addition to the G11778A mutation. Previously, the co-occurrence of the G11778A and A1555G mutations in mtDNA has been reported in a Chinese family with a high penetrance of LHON.15 The present case exhibited not only the primary heteroplasmic G11778A mutation in ND4 but also a homoplasmic T3394C mutation in ND1. The T3394C mutation, which is characterized as a mutation associated with LHON, is not common in LHON patients, and the significance of this mutation in LHON pathogenesis is not fully understood. However, since ND1 is a component of mitochondrial respiratory complex I, and the mutation in ND1 may thus decrease the activity of mitochondrial respiratory enzymes, it has been suggested that mtDNA ND1 is a hot spot for LHON mutations.16 In addition, the T3394C mutation has been reported in cases with diabetes mellitus, myopathy, encephalomyopathy, cardiac arrhythmia, and Fraser syndrome.17-21
While the patient's mother had the same mutation in her mtDNA, and brain MRI disclosed mild atrophy in the brain stem and cerebellum, LHON-plus symptoms were not observed. These results suggest that LHON is not a monogenetic disease and that it requires additional factors to trigger its phenotypic expression. Since G11778A alone seldom induces disturbance in the CNS, coexistence with T3394C may play an important role in the appearance of olivocerebellar degeneration. Since the G11778A mutation was heteroplasmic, we carried out semiquantitative analysis of the degree of the G11778A mutation. The results revealed 92% and 70% heteroplasmic mutations of G11778A in the proband (III-1) and his mother (II-2), respectively. This suggests that the phenotypic severity in this family is associated with the degree of G11778A mutation in the mtDNA, and coexistence of the homoplasmic T3394C mutation may be a trigger of olivocerebellar degeneration.
The combination of G11778A and T3394C mutations has been associated with the penetrance of optic neuropathy in some Chinese cases.22 However, the cases reported previously exhibited a homoplasmic G11778A mutation and a homoplasmic T3394C mutation, and the neurological examinations were not described in detail. Our report suggests that the combination of heteroplasmic G11778A and homoplasmic T3394C mutations in the mtDNA induces not only optic neuropathy but also degeneration of the olivocerebellar projections.


 XML Download
XML Download