

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Preclinical Alzheimer's Disease
A definite diagnosis of Alzheimer's disease (AD) requires histopathological evidence via autopsy or brain biopsy. Senile plaques (SPs) and neurofibrillary tangles (NFTs) are the main neuropathological hallmarks of AD,1 and these are used as criteria for making the pathological diagnosis of AD.2-4 However, lesions such as SPs and NFTs can be also observed in cognitively normal elderly people.5 SPs are extracellular deposits of amyloid in the gray matter of the brain, and NFTs are abnormal tau aggregations within the neurons of the brain. Amyloid deposition has been estimated to begin perhaps 10 years or more prior to any clinical signs of dementia. This deposition progresses with time until it reaches a plateau.5 In addition, abnormal tau aggregation, which appears to begin independently during normal aging and in the early stages of AD, is further accelerated by the concomitant amyloid pathology.5,6 That is, by the time the clinical dementia just starts to be detectable, densities of SPs and NFTs sufficient to meet the pathological criteria for a diagnosis of AD have already been established.7 These findings have led to the concept of preclinical AD. The time gap between the neuropathological changes and the clinical cognitive changes of AD is called preclinical AD.5,8 The onset of very mild dementia is related to synaptic and neuronal loss that is presumed to eventually result from the pathological progresses underlying the formation of SPs and NFTs (Table 1).9 There is little or no neuronal loss in aging or preclinical AD, but there is substantial loss in very mild AD. Recent autopsy data have confirmed that gross cerebral atrophy, indicating the loss of synapses and neurons, is the pathological substrate of the cognitive impairment in AD patients.10
The clinical course of AD commences with a presymptomatic or preclinical phase. With preclinical AD, it is assumed that the AD pathologic process in cognitively normal elderly people results in progressive neurodegeneration, and that affected individuals will develop symptomatic AD if they live long enough, although the time to symptomatic AD may be influenced by brain and cognitive reserve,11,12 and by other factors that are currently unknown. Next is symptomatic AD, which is further divided into two phases according to the clinical severity. The second phase is thus a prodromal phase of AD, and is commonly known as mild cognitive impairment (MCI),13 and the third phase in the evolution of AD is dementia, which is defined as impairments in multiple domains that are severe enough to produce loss of function.
Biomarkers of AD
Research on the biomarkers of AD has made great progress, and especially with regard to the use of biomarkers as diagnostic and prognostic tools. To date, cerebrospinal fluid (CSF) assays of amyloid β (Aβ) and tau, and amyloid imaging with Pittsburgh compound B (PIB)-positron-emission tomography (PET) are considered to be molecular biomarkers for AD. Fluorodeoxyglucose (FDG)-PET studies of brain metabolism and MRI of brain structure are downstream markers of the presence of AD pathology. These findings are also used to support the revised National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria for the research diagnosis of AD.14 Moreover, there is sufficient evidence that these biomarkers can be used to predict who will progress from MCI to AD or from normal to MCI or AD.15,16
Powerful, unbiased screening approaches utilizing highly sensitive proteomics techniques and novel applications of mass spectrometry are now producing a revolution in biomarker discovery. The use of genetics in combination with biomarkers will likely provide more diagnostic and prognostic information than the use of biomarkers alone. However, this review focuses on the most widely studied and well validated fluid and imaging biomarkers, as mentioned briefly above. The positive links between these biomarkers and AD pathology are summarized (Table 2), and studies searching for the antecedent biomarkers in preclinical AD are introduced.
Fluid biomarkers
CSF Aβ
Aβ peptides are generated from the cleavage of amyloid precursor protein (APP) by β- and γ-secretases.17 Aβ40 and Aβ42 are the most common isoforms among those that are 39-43 amino acid residues long.17 Levels of total CSF Aβ and Aβ40 do not differentiate individuals with AD from controls,18,19 although it has been shown that CSF Aβ40 levels are decreased in individuals with cerebral amyloid angiopathy.20 However, levels of CSF Aβ42 are commonly decreased in patients with AD,21 possibly because Aβ42 is the main component of AD plaques,22 which may function as sinks or traps for Aβ42,23 thus decreasing the amount of Aβ42 cleared from the brain to the CSF. However, decreased CSF Aβ42 levels have also been reported in non-AD dementias such as frontotemporal dementia (FTD), vascular dementia, Creutzfeldt-Jakob disease, and dementia with Lewy bodies.24-26 A possible limitation of AD studies that have used CSF Aβ as a marker is the lack of standardization for Aβ quantification. Furthermore, little is known about the influences of normal aging on CSF Aβ turnover and clearance. An important consideration is the normal circadian variability of the CSF Aβ40 and Aβ42 levels,27 indicating that differences in the time of day at which CSF is collected may contribute to variability of results. Levels of CSF Aβ42 do not correlate well with the disease duration or severity,28 a finding that is consistent with results from PIB-PET studies showing that amyloid retention does not change appreciably in symptomatic AD.29 These findings suggest that amyloid pathology occurs very early in the disease process and may have stabilized by the time the clinical signs of dementia appear.
Several studies have investigated CSF Aβ42 levels in conjunction with those of tau. Sunderland et al.28 assayed 131 AD patients and 72 controls and performed a meta-analysis of 17 studies on CSF Aβ42 levels and 34 studies on CSF tau levels. In their own cohort, they observed significantly lower mean CSF Aβ42 level and higher CSF tau levels in the AD patients compared to controls. The results of the meta-analysis were similar, with a difference in the two levels between the AD patients and the controls.
While plasma Aβ42 levels are increased and those of Aβ40 decreased in individuals with autosomal dominant, familial AD,30 most groups have reported no difference in the plasma Aβ levels between individuals with sporadic AD and controls.31-33 The diagnostic utility of plasma Aβ has been further limited by its short half life in the plasma (typically 5-15 min), its presence in very low concentrations, and by the additional peripheral sources of Aβ production and clearance, which can be influenced by confounding factors (such as renal function).
CSF tau
Many studies found that the CSF tau levels were increased in AD patients.19,21,25,28,34,35 Increased levels of CSF tau are markers of neuronal injury from multiple causes and can be seen in other neurodegenerative disorders such as FTD, stroke, and Creutzfeldt-Jakob disease.35 Increased CSF tau is not specific for AD, but does correlate with clinical disease severity, with higher concentrations associated with greater cognitive impairment in individuals with normal cognition and in patients with AD.36
In AD, tau undergoes abnormal hyperphosphorylation. As a result, it is unlikely to be able to bind and stabilize microtubules, possibly leading to axon degeneration, and so the increase in CSF tau in AD patients would be due to the release of tau from degenerating neurons and its subsequent diffusion into the CSF.37 Studies have shown consistently that phosphorylated tau (p-tau) offers equivalent and possibly better diagnostic utility for AD than total tau. In contrast to the total tau, p-tau is not increased secondary to acute brain injury, which further increases its diagnostic specificity.
Enzyme-linked immunosorbent assays have been developed to recognize various phosphorylated epitopes.38 The results of studies comparing the diagnostic accuracy of different phosphorylation sites such as p-tau231, p-tau181, and p-tau199 suggest that all three are equally effective in differentiating AD patients from controls.39 While p-tau231 appears to provide diagnostic specificity for AD and to improve the differentiation between AD and FTD,40 there is evidence that p-tau181 improves the differentiation between AD and dementia with Lewy bodies.39
Imaging biomarkers
Structural MRI
Neuropathological studies have documented an abundance of NFTs and significant neuronal loss in the hippocampus and entorhinal cortex of AD patients.1,8 Atrophy of the medial temporal areas, including the hippocampus and entorhinal cortex, has been observed on brain MRI of AD patients.41,42 Structural MRI thus offers an indirect marker of neuronal atrophy and loss of brain tissue, which are hallmarks of the neurodegenerative pathology of AD. Meta-analyses have confirmed the ability of MRI to distinguish AD subjects from controls, with volumetric studies of the medial temporal lobe and hippocampus having a sensitivity of 78-94% and specificity of 60-100%.43
In addition to discriminating AD from controls, volume measurements of the entorhinal cortex and hippocampus have been shown to discriminate MCI patients who later progress to AD dementia from those who do not.44,45 Longitudinal studies have demonstrated that the rate of whole-brain atrophy increases more in early-AD patients than in controls.46,47 In addition, a recent study found that the rate of ventricular volume expansion predicted future MCI in nondemented cohorts that were followed for up to 15 years, and that this rate further accelerated years prior to the diagnosis of MCI, suggesting that this measurement is also useful as an antecedent biomarker.48
As a surrogate of neurodegeneration, NFT formation, and neuronal and synaptic loss,49,50 MRI may correlate better with cognitive function than CSF biomarkers in cross-sectional and longitudinal studies.51,52 Structural markers that reflect rates of atrophy can also be useful for monitoring disease progression and severity. Volumetric measures of the hippocampus are already being employed as secondary endpoints in several pharmacologic trials, and in the near future, these measurements may be approved as surrogate endpoints and secondary outcome variables in trials of potential disease-modifying therapies.
Metabolic FDG-PET
PET has been employed in many AD studies to examine the regional cerebral metabolic rate for glucose (rCMRGLc) using 18F-2-deoxy-2-fluoro-D-glucose as a marker. A reduction of glucose metabolism, as seen on PET in the bilateral temporal parietal regions and in the posterior cingulate, is the most commonly described diagnostic criterion for AD.53 A meta-analysis of nine studies revealed that the pooled sensitivity and specificity were both 86% for temporoparietal hypometabolism when discriminating AD patients from healthy controls.54
In addition, a decreased rCMRGLc in the hippocampus was found to be indicative of who will progress to MCI among a group of cognitively normal individuals.55 Similarly, in longitudinal studies of individuals with MCI, the individuals who progressed to AD dementia had significant rCMRGLc reductions in the hippocampus and temporal neocortex as compared to those who did not progress.56 Consistent with the knowledge that the entorhinal cortex is one of the earliest affected areas in AD, hypometabolism therein has also been shown to accurately predict a decline to MCI or a Clinical Dementia Rating (CDR)57 score of 0.5 (CRD 0.5) with a sensitivity of 83% and a specificity of 85%.58 Jagust et al. reported that FDG-PET correlated well with CSF Aβ42 levels and with cognitive function.59
Molecular PIB-PET
PET imaging using 11C-labeled PIB {2-[4'-(methylamino) phenyl]-6-hydrobenzothiazole} ligand has been one of the major diagnostic tools in AD.60 PIB binds with high affinity and high specificity to fibrillar Aβ in neuritic plaques and cerebral amyloid angiopathy.29 In AD patients, PIB retention is increased in the frontal, parietal, temporal, and occipital cortices, and in the striatum, and studies have consistently shown that nearly all patients diagnosed with dementia of the Alzheimer type (DAT) are PIB positive [PIB(+)].29,61
Interestingly, a longitudinal study of AD patients who were taking cholinesterase inhibitors and/or the NMDA (N-methyl-D-aspartic acid) antagonist found that PIB retention did not change over a 2-year period of follow-up, although the cortical rCMRGLc decreased.62 This suggests that amyloid deposition reaches a maximum early in the course of AD, and indeed several studies have found that MCI subjects have PIB uptake in the same range as that of AD patients.29,63 In one study, initial PIB retention was predictive of disease progression over the next 2 years.29
Recent results from clinical trials of the 18F-labeled tracer 18F-AV-45 are promising for the next generation of amyloid imaging. It has several unique characteristics that make it suitable for Aβ plaque imaging in the human brain: excellent binding affinity, highly selective for Aβ plaque labeling, excellent brain penetration, and rapid kinetics in animal studies.64 Studies suggest that 18F-AV-45 is a sensitive marker for the presence of amyloid in cortical gray matter in elderly individuals, and can differentiate between groups of subjects with AD, MCI, and normal cognitive function.65 At the International Conference on Alzheimer's Disease in 2010, Clark et al. reported their phase 3 histopathology data (http://www.alzforum.org/new/detail.asp?id=2507). Similarly to the previous analysis of six autopsy cases,66 Clark and his colleagues found a near-perfect correlation between PET imaging using the new tracer and amyloid load measured postmortem in the same patients. In that study they tested florbetapir (formerly 18F AV-45) in 35 people who were expected to die within 6 months. Of the 19 subjects who met National Institute on Aging-Reagan criteria for AD pathology, all but 1 were amyloid-positive on PET, as judged by visual interpretation (97% accuracy), and all 19 came out positive on standard uptake value ratio quantification of PET data (100% accuracy). For both PET analysis methods, all 16 who lacked postmortem AD pathology were also amyloid-negative by live brain imaging, giving the tracer a specificity of 100% in this study.
Search for antecedent biomarkers of preclinical AD
Biomarkers to detect preclinical AD
Since amyloid deposition is known to precede clinical signs of dementia, PIB-PET may facilitate the early detection of amyloid during preclinical AD. In fact, up to 30% of cognitively normal elderly people demonstrate substantial PIB retention in the cortex by their mid-70s, and this PIB retention is similar in extent to that of patients with mild-to-moderate AD.67 These findings were expanded further by Fagan et al.,34,68 who reported an inverse relationship between CSF Aβ42 levels and brain amyloid load, as measured by PIB-PET. These findings suggest that a low CSF Aβ42 level is an excellent marker of amyloid deposition, independent of clinical status. PIB binding and CSF Aβ42 levels did not consistently correspond with clinical diagnosis. Cognitively normal (CDR 0) individuals who are PIB(+) with a low CSF Aβ42 level have cerebral deposits of amyloid in the absence of cognitive impairment (i.e., preclinical AD). These observations suggest strongly that the CSF Aβ42 level is a highly sensitive and specific marker for the presence or absence of amyloid in the brain (regardless of the clinical diagnosis), and so it may serve, either alone or in combination with PIB-PET, as an antecedent biomarker of AD.68 This important finding suggests that the inadequate sensitivity and specificity of CSF Aβ42 for distinguishing between clinical groups reflects contamination of the control group with preclinical cases of AD, and perhaps misdiagnoses of non-AD dementias in the DAT group.
In addition to decreased Aβ42 levels, CSF levels of tau (and the specific p-tau species) are increased in individuals with AD. There are significant overlaps in the tau levels as well as the Aβ42 levels between AD patients and controls. Aggregation of Aβ plays a necessary part in AD, and especially in the preclinical phase of the disease. Aggregation of the microtubule-associated protein tau begins in cognitively normal individuals and appears to correlate with neurodegeneration. CSF tau elevation can be observed in cognitively normal individuals and may mark the transition from cognitive normality to symptomatic AD. In preclinical AD, CSF tau levels are correlated with the amount of amyloid deposition.6
Preclinical AD is not benign
It has been shown that elevated amyloid burden, as measured by PIB-PET or CSF Aβ42 levels, is associated with longitudinal cognitive decline and regional brain atrophy. In a longitudinal study of cognitively normal adults, Morris et al.69 observed that cognitively normal individuals who later progress to CDR 0.5 DATqqq initially had a higher PIB uptake. This finding suggests that cortical amyloid is predictive of future cognitive decline and symptomatic AD. In addition, reduced CSF Aβ42 levels were associated with brain atrophy in cognitively normal individuals, but not in patients with AD.70,71 Preclinical AD is not benign, and Aβ aggregation seems to drive neurodegeneration in the preclinical phase.6
The ratios of CSF tau/Aβ42 and p-tau/Aβ42 in cognitively normal individuals strongly and significantly predict progression to a CDR >0 or MCI. Li et al. reported that over a follow-up period of 42 months, all of those subjects who converted to MCI had elevated CSF tau/Aβ42 ratios, while none of them converted among those with a normal ratio.72 It appears that cognitively normal elderly people with high ratios have already developed Aβ deposition and neurodegeneration, and so this most likely represents preclinical AD. In a study by Fagan et al., 70% of those subjects with high ratios (as compared to only 10% of those subjects with a normal ratio) converted from CDR 0 to CDR >0 after 3-4 years.34 In that study, the CSF tau/Aβ42 and p-tau/Aβ42 groups did less well, and the levels of plasma Aβ42 did not correlate with PIB status. This observation suggests that the CSF Aβ42 level decreases with amyloid deposition, and that amyloid plaques act as a sink.
The time course of biomarker abnormalities during preclinical AD
Brain amyloid can be assessed based on represented as reductions in the CSF Aβ42 levels and increased PIB retention. Elevated CSF tau is thought to be a biomarker of tau-mediated neuronal injury and neurodegeneration. Decreased FDG uptake on PET in the temporoparietal area is a biomarker of AD-related synaptic dysfunction, and brain atrophy seen on structural MRI and involving the medial temporal lobe is a biomarker of AD-related neurodegeneration. The fluid and imaging biomarkers parallel the pathophysiological sequence of AD and are linked with AD pathology. Abnormal accumulation of Aβ42 in oligomeric forms is an early event in the pathophysiologic cascade of AD that ultimately manifests as cerebral deposits of Aβ. Through mechanisms that remain to be elucidated, it is postulated that the accumulation of Aβ leads to synaptic dysfunction, neurodegeneration, and eventually neuronal loss. Although it is further accelerated by the concomitant amyloid pathology, abnormal tau aggregation also begins independently during normal aging.5,6
Preclinical AD is characterized by significant Aβ deposition and lesser degrees of tau aggregation, with minimal neuronal loss. The biomarkers related to amyloid plaques become abnormal first, and a substantial amyloid load accumulates prior to the appearance of clinical dementia. Decreases in CSF Aβ42 levels may precede amyloid retention, as detected by PIB-PET, signifying what is perhaps the first evidence of AD pathology in cognitively normal individuals.68,70 A case report of the clinical, cognitive, and CSF markers of AD in an individual who progressed from cognitive normality to early symptomatic AD suggests that changes in these biomarkers precede the detection of cerebral fibrillar amyloid using PIB-PET.73 Nonfibrillar cerebral Aβ deposits or diffuse SPs are already pathognomonic, and they are not benign.
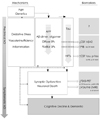
While amyloid and tau pathologies in preclinical AD are inevitably associated with some degree of neuronal, axonal, and synaptic loss, and neuronal injury, it is only after a threshold of neuronal loss has been reached in specific neocortical regions that clinical signs of dementia appear. These markers appear to correlate better with clinical impairment than the amyloid load. Structural MRI is the last biomarker to become abnormal. However, MRI retains a closer relationship with cognitive performance later into the disease than other biomarkers (Fig. 1).
The adult children study and the dominantly inherited alzheimer network
Perhaps the most important role of biomarkers, and the most needed at present, is the identification of individuals who are cognitively normal but who have evidence of AD pathology (i.e., preclinical AD). Such individuals can be identified with antecedent biomarkers such as the CSF Aβ42 and tau, and with PIB, and they are the most likely to benefit from future disease-modifying therapies.
There have been many trials to establish validated antecedent biomarkers. The Adult Children Study, which is a longitudinal assessment of middle-aged to elderly cognitively normal individuals, is currently evaluating potential indicators of incipient disease through an analysis of cognition, personality, genetics, biomarkers, and neuroimaging in a group of normal healthy people aged between 45 and 74 years. The individuals have been stratified into those with a parent who had AD and those for whom neither parent had AD. All of the subjects are subjected to MRI, FDG-PET, and PIB-PET amyloid imaging, and psychometric testing with a follow-up every 2 years; CSF biomarker analyses are also being performed. In 241 cognitively normal participants, the PIB-assessed amyloid burden increased as a function of the 2 known risk factors for AD: age and apolipoprotein ε4 (ApoE4).74 In the study participants, PIB uptake increased according to age, and a group of cognitively normal ApoE4-positive older individuals (mean age in the late 50s) showed a mean decrease in CSF Aβ42, but no change in CSF tau relative to an age-matched group of ApoE4-negative individuals. Cerebral Aβ42 deposition is the pathobiological phenotype of ApoE4, and this increases as a function of age in preclinical AD patients.
The Dominantly Inherited Alzheimer Network will establish an international registry of mutation carriers and noncarriers from families with presenilin (PSEN) 1, PSEN 2, or APP mutations. The purpose of the Dominantly Inherited Alzheimer Network is to compare mutation carriers and noncarriers to determine the chronology and order of imaging and biomarker changes that predict symptomatic AD, to compare the clinical and pathological phenotypes of dominantly inherited AD with those of late-onset AD, and to maintain a publicly available resource of data and biospecimens. Presymptomatic carriers of AD mutations such as PSEN 1 and APP exhibit decreased CSF Aβ42 and increased CSF tau.75,76
These studies have compared candidate biomarkers between high- and low-risk groups, from which promising biomarkers may be obtained through immediate comparison of samples therefrom. The high-risk groups used for these kinds of studies could be defined by genetic variables, for example, presymptomatic individuals with familial AD mutations77 or carriers of ApoE4.78 Other risk factors could also be used to define risk groups, such as those with advanced age79 or a family history of AD.80
Conclusions
Aβ and tau as the main pathological substrates of AD have driven the search for the biomarkers of AD. Of course, the pathophysiology of AD involves many more processes than Aβ deposition and NFT formation. APP is cleaved by β-secretase and γ-secretase complexes. Once released in monomer form, Aβ may form oligomers that are neurotoxic. Aβ accumulates and aggregates to form plaques. Once Aβ has formed oligomers and amyloid deposits, microglial cells become activated and migrate toward the plaques. Astrocytes become reactive, and numerous inflammatory mediators, oxidative processes, and protein-folding activities are released. Dendrites and axons around the plaques become dystrophic due to a transportation defect. The brain metabolism changes as Aβ is deposited in the small or large vessel walls. Neuronal injury and synaptic loss develop in addition to formation of NFTs, and then neurons die (Fig. 1). Each of these changes may also cause alterations in the composition of the CSF and plasma, and these changes may be therapeutic targets for disease-modifying therapies. Biomarkers can lead to the early diagnosis of AD and can be used to detect preclinical AD. Convincing evidence has expanded the scope of AD research, and so new biomarkers have been included in the diagnostic criteria of the proposed revisions of National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (www.alz.org/research/diagnostic_criteria/). The operational research criteria suggest that we can diagnose preclinical AD with just the findings of Aβ accumulation according to PIB retention or a low CSF Aβ42 level, in addition to synaptic dysfunction and early degeneration such as hypometabolism seen on FDG-PET, cortical thinning or hippocampal atrophy on MRI, and elevated CSF levels of tau or p-tau. Moreover, these findings are used as surrogate outcome measures to predict the time course of future cognitive decline. However, we should distinguish between clinical criteria and the research criteria.
While there have been promising results related to determining the antecedent biomarkers of AD, the reliability and validity of biomarkers and definition of the cut-off values need to be established. Moreover, we should not neglect the neuropsychological assessments for making the diagnosis of early AD, although clinical evaluations, by definition, will not identify the presence of preclinical disease. It is important to remember the potential that behavioral markers hold for AD. A study observing the transition from healthy aging to symptomatic AD found a sharp inflection point followed by an accelerating decline in multiple domains of cognition (not just in memory) during the preclinical period of AD when there was insufficient cognitive decline to warrant a clinical diagnosis with the aid of conventional criteria.81 Additional longitudinal studies of older individuals could provide more information, and perhaps by combining biomarkers with other measures that can sensitively detect very subtle cognitive decline.
The long preclinical phase has profound implications for AD therapeutic strategies. Since potential intervention with disease-modifying therapies may provide the greatest chance of preserving normal cognition, it will be critical to identify individuals with preclinical AD before the development of cognitive deficits and concomitant neuronal loss. Thus, there may be a paradigm shift in AD from cure to prevention. The hope is that in the future, AD will be managed in the way cardiovascular disease is handled now. Physicians will use lifestyle factors and diagnostic measures to define the risk of AD in their patients, followed by manipulation of their diet, lifestyle, and medications to delay or prevent the symptomatic onset of this disease.


 XML Download
XML Download