

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Progressive external ophthalmoplegia (PEO) is one of the most common manifestations of mitochondrial disease, either as a part of a multisystemic complex disorder or as a predominant feature of exclusive mitochondrial myopathy. Many cases of PEO are caused by the mutation of mitochondrial DNA (mtDNA). A single heteroplasmic deletion of mtDNA is a frequent cause of sporadic PEO, being responsible for approximately 90% and 50% of cases of Kearns-Sayre syndrome and PEO, respectively.1 In other patients who show maternal inheritance pattern, PEO is associated with various point mutations of mtDNA, of which A3243G is the most frequent.2 Yet another group of PEO patients display autosomal dominant or recessive inheritance patterns that are usually associated with multiple mtDNA deletions or depletions that arise secondarily to nuclear gene defects.3 The responsible genes include POLG, which encodes the catalytic subunit of DNA polymerase γ; SLC25A4, which encodes the mitochondrial adenine nucleotide translocator ANT1; C10ORF2 (also known as PEO1), which encodes T7 gene-4-like protein with intramitochondrial nucleoid localization (TWINKLE), a mitochondrial protein that is structurally similar to the phage T7 primase/helicase; POLG2, which encodes the p55 accessory subunit of DNA polymerase γ; OPA1, which encodes optic atrophy 1; and RRM2B, which encodes the small subunit of the ribonucleotide reductase p53R2.4-8
While a few previous studies have documented a single large-scale mtDNA deletion in patients with PEO and Kearns-Sayre syndrome,9-11 there are no reported cases of PEO associated with a nuclear gene mutation in Korea. In this study we analyzed two Korean patients with autosomal dominant PEO (ad-PEO) to identify the nuclear gene defects that underlie their clinical phenotypes and pathology.
Methods
Using our muscle biopsy database, we identified ten patients with PEO in whom mitochondrial myopathy was confirmed pathologically. Among the patients, two unrelated patients who had a family history compatible with ad-PEO were selected for the study. For DNA analysis, written informed consent was obtained from the patients, and the study was approved by the Pusan National University Hospital Institutional Review Board. The DNA was extracted from a biopsied muscle sample from each patient and subjected to a long-range polymerase chain reaction (PCR) protocol for the amplification of mtDNA, as described previously.12,13 After exclusion of a single large-scale rearrangement, PCR amplification of all coding exons and intron-exon boundaries of POLG, SLC25A4, C10ORF2, and POLG2 was performed (primer pairs and reaction conditions are available on request). Each PCR product was directly sequenced using a BigDye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and a 3730×l DNA analyzer (Applied Biosystems). The obtained chromatograms were compared with the standard DNA sequence of each gene, and the aberrant sequences were searched in The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/).14
Results
DNA analysis
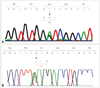
Upon long-PCR amplification of mtDNA, no single large-scale mtDNA deletion was observed in either patient, and multiple mtDNA deletions were observed in one patient (patient 2). Subsequent sequence analysis revealed heterozygous missense mutations in each patient: c.311A>G (p.Asp104Gly) on exon 2 of SLC25A4 in patient 1, and c.1435G>A (p.Glu479Lys) in exon 2 of C10ORF2 in patient 2 (Fig. 1). No other mutation was identified in any other gene investigated.
Clinical features
Patient 1 (SLC25A4 p.Asp104Gly mutant)
The patient was a 65-year-old female who presented with progressive bilateral ptosis. The ptosis had begun about 50 years previously, at school age, with a subsequent slow progression. The patient reported no diplopia, visual disturbance, hearing loss, or gait difficulties. She had presented with hypertension and a cerebral infarction at the age of 55 years, which had produced mild right hemiplegia. The patient's family history was significant for the autosomal dominant inheritance pattern of PEO; starting from school age, at least 15 members from 4 generations of her family were affected (Fig. 2A). An examination revealed bilateral ptosis, partial limitation of extraocular movement in all directions, and mild facial weakness. The patient had normal-sized, round pupils with prompt light reflexes; the optic fundi were normal. No other cranial nerve abnormalitiesincluding hearing loss, dysarthria, dysphagia, neck flexor weakness, and tongue atrophy-were observed. The patient's individual limb muscle power was preserved, except for the mild right hemiparesis that had developed 10 years previously (at an age of 55 years) due to cerebral infarction. The patient's cognitive, sensory and cerebellar functions were normal, and no involuntary movement was observed. Deep tendon reflexes were absent bilaterally in the biceps, triceps, and ankles, and were reduced bilaterally in the knees. A Babinski sign could be evoked on the right side. Tests including chest roentgenogram, electrocardiogram, complete blood count, liver and renal function test, serum glucose, electrolytes, creatine kinase, lactic acid, pyruvic acid, and thyroid function test were all within normal limits. Needle electromyography revealed fibrillation potentials, positive sharp waves, and motor unit potentials with small amplitudes and short durations in all limb muscles tested. Nerve conduction evaluation yielded normal findings. A muscle biopsy of the left biceps muscle revealed numerous ragged red fibers with focal cytochrome oxidase deficiency (Fig. 3A-D).
Patient 2 (C10ORF2 p.Glu479Lys mutant)
This patient was a 47-year-old male with progressive bilateral ptosis, ophthalmoplegia, dysarthria, and dysphagia. The patient first developed bilateral ptosis, dysarthria, and swallowing difficulty at ages of 35, 40, and 44 years, respectively. The patient also complained of myalgia and paresthesia affecting the lower legs 2 years before this visit, and had a significant 10-year weight loss from 57 to 40 kg. The patient's family history was significant for ad-PEO; his father, an elder sister, and an elder brother had progressive ptosis, dysarthria, and dysphagia that had begun in childhood (Fig. 2B). A physical examination revealed that the patient had a very thin musculature, with a weight of 40 kg and a height of 170 cm. Neurological examination revealed bilateral ptosis, nearly complete ophthalmoplegia, masseter and temporalis muscle atrophy, facial weakness, moderate sensorineural hearing loss with tinnitus, dysarthria with nasal voice, dysphagia, and neck flexor weakness; the optic fundi were normal. The patient's limb muscle power was preserved except for a mild elbow flexor weakness, which was grade 4 on the Medical Research Council scale. The patient had a reduced vibration sensation in the lower extremities, and hypoactive deep tendon reflexes in the limbs. No other neurological abnormalities were observed. On laboratory examination, the patient displayed a mildly elevated serum creatine kinase level (342 U/L; normal range, 5-217 U/L). An electrocardiogram showed a nonspecific ST-segment elevation in the inferior and anterior walls. A chest roentgenogram suggested previous pulmonary tuberculosis; no evidence of cardiomegaly was observed. The results of other laboratory tests including serum lactate/pyruvate, complete blood count, liver and renal functions, electrolytes, blood sugar, and thyroid function were unremarkable. A nerve conduction study revealed reduced compound nerve action potential amplitudes bilaterally in the sural nerves and mildly slow sensory nerve conduction velocities in the median and ulnar nerves. Needle electromyography revealed mild neurogenic changes in the limb muscles. The overall electrodiagnostic findings were compatible with axonal-type sensorimotor peripheral polyneuropathy rather than myopathy. A muscle biopsy performed on the left biceps muscle revealed many ragged red fibers with partial deficiencies of cytochrome oxidase activities (Fig. 3E-H).
Discussion
The nature of multiple mtDNA deletions in PEO with Mendelian inheritance has been evaluated previously using single-muscle-fiber PCR analysis, which showed that each fiber has one type of deletion that is different from each cell, suggesting that each mtDNA with deletions originates in a mitochondrion and expands in postmitotic cells.15 PEO with multiple mtDNA deletions is a genetically heterogeneous entity caused by mutations of nuclear genes that are involved in the replication and maintenance of mtDNA. Many ad-PEO families carry a heterozygous mutation in one of six genes (POLG, POLG2, SLC25A4, C10ORF2, OPA1, or RRM2B), while many autosomal recessive PEO families carry POLG mutations. Previous studies showed that POLG, SLC25A4, and C10ORF2 are three major genes that are mutated in ad-PEO, among which POLG is the most frequently mutated, with mutations SLC25A4 and C10ORF2 being uncommon.16 OPA1 was not analyzed in our study because the OPA1 mutant is always associated with optic atrophy,17 which was not a feature of our patients. Another gene, RRM2B, was not analyzed, since ad-PEO with a RRM2B mutation is considered extremely rare, and only two families with the same p.R327X mutation have been reported to date.8
SLC25A4 encodes ANT1, or ADP/ATP translocator, which is the most abundant protein in the mitochondrion. ANT is located in the inner mitochondrial membrane as a 30-kD homodimer, where it forms a gated pore through which ADP is exchanged with ATP. ANT1 is one of the three isoforms of ANT that is predominantly expressed in heart and skeletal muscle.18 Mutations of ANT1 are associated exclusively with ad-PEO, and only six missense mutations have been reported thus far.14 The mutation identified in our study (p.Asp104Gly) has been reported previously in a Japanese family with a late-onset pure PEO phenotype, and has been well characterized as a pathogenic mutation.15 Although the exact mechanism by which ANT1 mutations produce multiple mtDNA deletions leading to ad-PEO is not yet clearly understood, several mechanisms including disturbed intramitochondrial deoxyATP concentration, disturbed mitochondrial permeability transition pore opening caused by structural defect of ANT1, and increased oxidative stress associated with ANT1 dysfunction have been suggested.4
C10ORF2 encodes TWINKLE; it is a primary mtDNA helicase with nucleotide hydrolase activity and is localized to mitochondrial nucleoids.5 TWINKLE interacts functionally with the β subunit of polymerase γ,19 and is stimulated by the mitochondrial single-stranded DNA-binding protein20 to form a replication unit of mtDNA.21 Thus, mutation of C10ORF2 produces defects in both the maintenance of mtDNA copy number and the integrity of mtDNA, which leads to multiple deletions of mtDNA and, consequently, disease. Clinically, POLG and C10ORF2 mutants share similar phenotypes including PEO, myopathy, ataxia, sensory axonal neuropathy, and Parkinsonism, and this is probably related to the close functional interactions between polymerase γ and TWINKLE.22 More than 29 different mutations have been identified, most of which are missense mutations involving exon 1 or 2.22 The presently identified C10ORF2 mutation (p.Glu479Lys) is also located in exon 2, and was previously reported as a de novo mutation from a young Italian female with early-onset PEO and a stationary clinical course.23 However, our patient showed different clinical features: clear family history of autosomal dominant inheritance, onset in the mid-thirties with clinical progression, and PEO plus other additional features including dysphagia, dysarthria, proximal myopathy, and axonal-type sensorimotor polyneuropathy. Although these findings may indicate the phenotypic heterogeneity of this particular mutation, further accumulation of clinical and genetic data are needed to clarify this issue.
Two known missense mutations have been identified in this study in two Korean patients with ad-PEO: one in SLC25A4 and the other in C10ORF2. The clinical phenotype of the SLC25A4 p.Asp104Gly mutant was similar to that described previously, while the C10ORF2 p.Glu479Lys mutant phenotype was quite different. Further molecular biological and clinical genetic studies employing larger numbers of patients are needed to establish the clear pathogenetic mechanisms and genotype-phenotype correlations in ad-PEO.


 XML Download
XML Download