

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
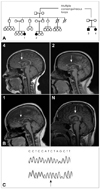
The hereditary spastic paraplegias are a clinically and genetically heterogeneous group of disorders. Recessive HSP tends to be complicated with additional features such as ataxia, neuropathy, seizures, and cognitive decline.1 Recessive mutations in the spatacsin gene underlie spastic paraplegia type 11 (SPG11), the most-common type of recessive HSP in which affected individuals exhibit a thin corpus callosum (TCC) in addition to progressive spasticity, ataxia, neuropathy, seizures, cognitive decline, and abnormal eye signs.2 We describe herein a large family originally from Gujarat in North India (Fig. 1A) with progressive spastic paraplegia in whom we identified homozygous mutations in the spatacsin gene associated with SPG11. We report a relationship between disease severity in affected members, the addition of parkinsonian features in the most-severe case, and radiological correlation of the disease, in particular TCC and cerebellar atrophy.
Case Report
This family was referred to the HSP and rehabilitation clinic at The National Hospital for Neurology and Neurosurgery in London, UK. The family had come to London over 30 years ago and was originally from Gujarat in North India. After the establishment of consanguinity, it was noted that the four affected family members had presented in adolescence with varying degrees of progressive spastic paraplegia and cognitive impairment. Additional features included mild muscle wasting. The clinical features and the MRI findings were similar in each individual, but varied widely in severity (Fig. 1B). In addition, the findings of nerve conduction studies and electromyography were normal in all four individuals.
Patient 1 was the proband and the most-severely affected member. Her motor milestones were delayed, and in particular her walking. She had developed progressive clumsiness, poor coordination, unsteadiness on her feet, and cognitive impairment from the age of 8 years. She had always been poor at sports, and described difficulty walking and frequent falling over in her teenage years. Examination at the age of 29 years demonstrated bilateral ptosis, significant cerebellar ataxia, and spastic dysarthria; she also had hypometric saccades with broken pursuit movements; tone was raised in her upper limbs and profoundly increased in her lower limbs. In addition, she exhibited parkinsonian features with a stooped, festinant gait, a hypomimic face, bradykinesia, and cogwheeling in her upper limbs. She had a positive glabellar tap and limitation of upward gaze.
Patient 2 had milder symptoms. From the age of 10 years she had developed progressive gait difficulties, stiffness, and clumsiness. Examination at the age of 22 years revealed a spastic quadriparesis, spastic dysarthria, and cerebellar ataxia. She also exhibited limitation of upward gaze and skew deviation of the left eye. She had only mild cognitive impairment.
Patients 3 and 4 are siblings, and cousins of patients 1 and 2. Patient 3's phenotype was almost identical to that of patient 2, while patient 4 was the least-affected member of the family, with symptom onset at the age of 15 years. Examination at the age of 18 years revealed normal upper limb tone, power, and reflexes. She exhibited only mild lower limb spasticity, but power was normal and her reflexes were not brisk. There was no evidence of cerebellar ataxia on examination. She exhibited no cognitive impairment.
Brain MRI demonstrated TCC and cerebellar atrophy (Fig. 1B), which correlated with disease severity in the affected members. In addition, patient 1 had an increased signal in the basal ganglia on T2-weighted MRI, perhaps corresponding to her parkinsonism. Analysis of spatacsin (KIAA1840) in all affected individuals identified a homozygous mutation, c.5769delT (Fig. 1C), which results in a truncated protein (p.Ser1923ArgfsX28), confirming the diagnosis of SPG11.2,3
Discussion
The affected members of this family with SPG11 exhibited varying degrees of phenotypic severity and the development with age of additional features such as cerebellar ataxia and cognitive impairment. Although the absence of these findings in all affected individuals is likely to be age related, other genes are also likely to influence the overall phenotype. Interestingly, patient 1 exhibited features of parkinsonism, which has only been described in the literature once before.4 This feature may be associated with more advanced disease, since few SPG11 cases have been assessed in their 30s. This is a potentially treatable manifestation that should be carefully assessed in patients.
The extent of TCC and cerebellar atrophy was correlated with phenotypic severity in the affected members of this family. The most-striking findings were observed in patient 1, who exhibited the most-severe phenotype, and the least atrophy was seen in patient 4, who had the mildest phenotype. The phenotypes of patients 2 and 3 were of intermediate severity. TCC is an important diagnostic feature in patients with autosomal recessive HSP, and seems to be correlated with disease severity. However, our observations are based on one family, and therefore need to be replicated in further affected families with patients of different ages and mutation types. Spatacsin pathogenic mutations have recently been identified in autosomal recessive juvenile amyotrophic lateral sclerosis, in the absence of cognitive impairment and TCC,5 suggesting that the clinical spectrum seen in spatacsin-associated disease is much wider than previously thought.
 XML Download
XML Download