

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
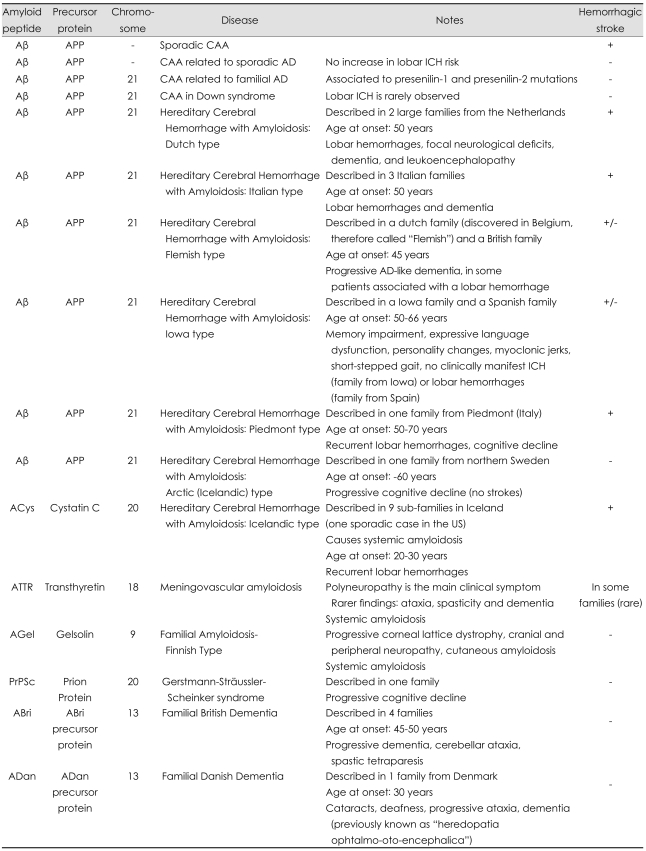
The term cerebral amyloid angiopathy (CAA) describes an heterogeneous group of biochemically and genetically diverse central nervous system (CNS) disorders. All these medical conditions share a characteristic morphological finding on pathological examination, i.e. amyloid fibrils deposited in the walls of small to medium-sized, blood vessels mostly arterial. In some instances amyloid deposits have been also observed in the capillaries of CNS parenchyma and of the leptomeninges. CAA mostly occurs in the sporadic form in the elderly, while rare familial forms occur in younger patients and are generally lead to more severe clinical manifestations. While more than 25 human proteins or have been found to form amyloid fibrils in vivo, only 7 have been described in CNS disorders,1,2 and all sporadic forms and most hereditary forms of CAA affecting the human brain are of the Aβ type (Aβ-CAA)(Table 1). In Aβ-CAA the action of β- and γ-secretases on the amyloid precursor protein (APP) lead to deposition of amyloid-β (Aβ) peptide, mirroring some aspect of Alzheimer's disease (AD) patophysiology.3
In this review we will present a summary of existing evidence regarding the epidemiology, genetics and pathogenesis of CAA, provide an overview of diagnostic tools available to clinical neurologists, as well as summarize current guidelines and recommendation for prevention and treatment of CAA-intracerebral hemorrhage (ICH)(See Appendix for literature Search criteria).
Historical Perspective
Vascular β-amyloid deposition in the central nervous system was first described by Gustav Oppenheim in 1909. Oppenheim found foci of necrosis in the brain parenchyma adjacent to hyalinized capillary walls in 6 of 14 brains of autopsied individuals with senile dementia and the pathological changes of AD.4 In 1938, Scholz published the first article focusing solely on cerebral vascular abnormalities now recognized as CAA.5 The observation that CAA is limited to the vascular media without adjacent parenchymal involvement was made in 1954 by Stefanos Pantelakis.6 He also described many of the hallmark pathological features of CAA: 1) preferential involvement of the small arteries and capillaries of the meninges, cerebral cortex, and cerebellar cortex; 2) topographical distribution favoring the posterior brain regions; 3) lack of staining of vessels in the white matter; 4) association with increased age and dementia; 5) lack of association with hypertension and arteriosclerosis; 6) lack of association with amyloidosis of the other organs. Over the following 20 years multiple case reports and small series suggested an association between CAA and lobar ICH. Okazaki and colleagues published a seminal article in 1979, clarifying the relationship between CAA and lobar ICH.7 They identified 23 consecutive cases of moderate to severe CAA from autopsies at Mayo Clinic (Rochester, MN). A history of lobar, multiple hemorrhages was very common in these patients. Fibrinoid degeneration of the vessel walls with microaneurysm formation was sometimes seen, as well as a double-barreled vessel wall appearance caused by cracking of the arterial media. Based on these findings, the authors concluded that CAA was an under-recognized cause of lobar ICH in the elderly, thus providing the foundation for further exploration of CAA and CAA-related lobar ICH (CAA-ICH).
Sporadic Cerebral Amyloid Angiopathy
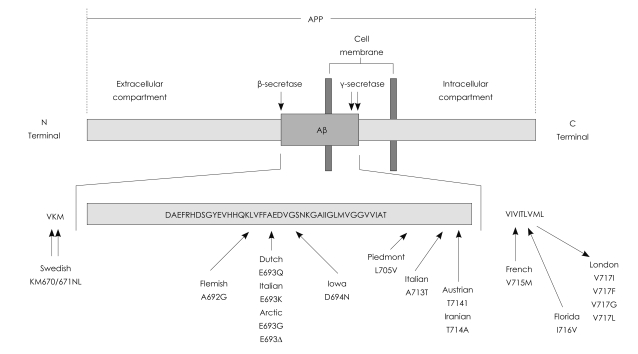
As previously mentioned (Table 1), sporadic CAA mostly occurs in the elderly because of Aβ deposition (Aβ-CAA). Aβ is a normally secreted, -4 kDa, 40 or 42 amino acids in length, proteolytic product of the 677-770 amino acid type 1 integral membrane protein referred to as the Aβ precursor protein (AβPP, or more commonly APP) encoded by the APP gene on chromosome 21.8-11 Generation of Aβ from APP requires two proteolytic events, a proteolytic cleavage at the amino terminus of the Aβ sequence referred to as β-secretase and a cleavage at the carboxyl terminus known as γ-secretase.
Epidemiology
CAA is a frequent pathological finding and a fairly common clinical entity in the elderly. As a detectable pathology (regardless of severity), cerebrovascular amyloid is present in approximately 10% to 40% of elderly brains and 80% or more in brains with concomitant AD.12 Even when taking only relatively advanced amyloid pathology in consideration, CAA remains a frequent finding. CAA pathology graded as moderate or severe (see below for pathology grading scores) was estimated to be present in 2.3% of 65 to 74 year olds, 8.0% in 75 to 84 year olds, and 12.1% in those over 85 years in analyses of brains from the Harvard Brain Tissue Resource Center. Of note, these figures were corrected for over-representation of AD referrals.13 An even higher estimate of 21% for the prevalence of CAA graded as severe emerged from other analyses of autopsied individuals aged 85 to 86.14 The role of CAA in cerebrovascular epidemiology is of course mostly related to the increased risk for lobar ICH. Indeed, estimates for the proportion of spontaneous hemorrhages in the elderly attributable to CAA range from 10% to 20% in autopsy series to 34% in clinical series.12,15
Pathogenesis
The 40-amino-acid-long Aβ (Aβ 1-40) is more soluble than the longer Aβ 1-42 and the two molecules differ in the distribution in brain and vessel walls. Aβ 1-40 tends to be the major form in the amyloid in artery walls in CAA, whereas Aβ1-42 is more prominent in the plaques in brain tissue.16,17 It is generally accepted that conformational transitions occurring in native soluble amyloid molecules increase their content in β-sheet structures, thus favoring the formation and deposition of more insoluble oligomeric structures. In turn, these deposits trigger a secondary cascade of events including, among others, release of inflammatory components, activation of the complement system, oxidative stress, alteration of the blood-brain barrier (BBB) permeability, and cell toxicity.18,19
While both deposited and soluble Aβ molecules are identical in their primary structure, they exhibit completely different solubility and tinctorial properties. Soluble Aβ forms undergo a change in conformation (via mechanisms that remains largely unknown) resulting in a predominantly β-sheet structure, highly prone to oligomerization, fibrillization and deposition. The identification of soluble Aβ species in the systemic circulation, brain interstitial fluid and CSF, together with the ability of the BBB to regulate Aβ transport in both directions, originally suggested plasma Aβ to be a potential precursor of deposited amyloid.20 However, lack of brain lesions in transgenic models exhibiting several fold increased in plasma soluble Aβ strongly argues against the sole contribution of circulating species to brain deposition.21
Since smooth muscle cells, pericytes and endothelial cells all express APP22 and isolated cerebral microvessels and meningeal blood vessels are able to produce Aβ23 the cerebral vasculature itself was therefore proposed as a possible source of cerebral Aβ. Nevertheless, the sole contribution of smooth-muscle cells to Aβ-CAA is made less likely by the existence of amyloid deposits in capillaries (which are devoid of smooth muscle) in CAA patients. In recent years, the hypothesis of a neuronal origin of Aβ and other amyloid proteins has therefore been gathering support.22,25-27 It has been proposed that amyloid produced by neurons is drained along the perivascular interstitial fluid pathways of the brain parenchyma and leptomeninges, depositing along the vessels under specific pathologic conditions.28,29
Since no evidence of increased Aβ production has been found in sporadic CAA, imbalance between Aβ production and clearance is generally considered a key element in the formation of amyloid deposits. The amphyphilic nature of Aβ precludes its crossing through the BBB unless mediated by specialized carriers and/or receptor transport mechanisms. These mechanisms control the uptake of circulating Aβ into the brain30-36 and regulate clearance.37-43 Among receptors involved, Receptor for Advance Glycation End-products actively participates in brain uptake of free Aβ at the vessel wall level.30 Other receptors are more relevant for the transport of Aβ complexed with other molecules: LRP-1 mediates transcytosis of Aβ-ApoE complexes contributing to rapid CNS clearance,41 whereas megalin mediates in the cellular uptake and transport of Aβ-ApoJ.36
The hypothesis of defective Aβ degradation, while less extensively studied as a possible mechanism for amyloid accumulation, should not be overlooked. Neprilysin, endothelin-converting enzyme, insulin-degrading enzyme, beta-amyloid-converting enzyme 1, plasmin and matrix metalloproteases are among the major enzymes known to participate in brain Aβ catabolic pathways.44-46 Reduced levels and/or activity of Aβ degrading enzymes favor Aβ accumulation, as documented in murine models, in which gene deletion of different proteases translate into increased levels of Aβ deposition.45,46
Genetics of sporadic CAA
The ApoE ε4 and ε2 alleles are the only genetic risk factors robustly associated with risk of developing sporadic Aβ-CAA.47 Interestingly ApoE ε2, which exerts a protective effect on AD risk, increases risk of ICH in Aβ-CAA patients.48,49 ApoE interacts with soluble and aggregated Aβ in vitro and in vivo and is therefore likely to be involved in both parenchymal and vascular amyloidosis.50-53 Further studies tested the role of human ApoE alleles on the formation of parenchymal and vascular amyloid. The presence of the ε4 allele led to substantial Aβ-CAA with only few parenchymal amyloid deposits. The ε3 allele, however, resulted in almost no vascular and parenchymal amyloidosis.54 In young mice, an increased ratio of Aβ 40 : 42 was observed in brain extracellular pools and a lower Aβ 40 : 42 ratio in CSF, suggesting that ApoE ε4 causes altered clearance and transport of Aβ within different brain compartments. These findings highlight again the importance of a high Aβ 40 : 42 ratio for the formation of vascular amyloid. Other genetic risk factors for Aβ-CAA have been investigated, but their role remains unclear, thus requiring additional research efforts.
Neuropathology
Characteristic findings
In hematoxylin-eosin stained sections severe CAA can be recognized by acellular thickening of blood vessel walls, but this finding is non-specific for CAA since it occurs in a variety of other disorders, including hypertensive angiopathy.55 In CAA, Aβ is deposited mainly as amyloid-β fibrils in close contact with smooth muscle cells.56,57 Non-fibrillar, monomeric and oligomeric Aβ was also demonstrated inside smooth muscle cells.56 Depending on the severity of CAA, Aβ depositions have been shown primarily in the abluminal portion of the tunica media, often surrounding smooth muscle cells, and in the adventitia. With increasing severity, Aβ infiltrates all layers of the vessel wall, which shows loss of smooth muscle cells. Finally, the vascular architecture is severely disrupted and "double barreling", microaneurysm formation, fibrinoid necrosis, and evidence of perivascular leakage may be seen.58 Even in very high degrees of CAA-related changes, endothelial cells are well preserved and usually not affected. Perivascular hemorrhages are frequent around blood vessels affected with CAA.
Several authors also reported CAA-associated inflammation/vasculitis.59,60 Two subtypes of CAA-associated inflammation have been described so far: a non-vasculitic form called perivascular infiltration, which is characterized by perivascular infiltration of the parenchyma by multinucleated giant cells and a vasculitic form called transmural granulomatous angiitis, which is characterized by inflammation of the vessel wall with the occasional presence of granulomas. Both pathologic forms can co-occur, suggesting at least a partial overlap in biological mechanisms.
Topographical distribution
CAA distribution is characteristically patchy and segmental. In one given histological slide there may be foci showing vessels with varying degrees of amyloid depositions adjacent to foci showing vessels without any amyloid deposition. This phenomenon might lead to an under-diagnosis of CAA in postmortem examination, as even in severe cases a given histological slide might not contain amyloid-laden blood vessels. It has been shown by many authors that CAA is most frequent in the occipital lobe, followed by either frontal, temporal or parietal lobes, respectively. Furthermore, the occipital lobe is not only the site most frequently affected with CAA but also most severely so. CAA is more rarely seen in the basal ganglia, thalamus, and cerebellum, while both white matter and brainstem are usually spared.61-63
Histological diagnosis
Histological diagnosis of CAA requires use of special staining for amyloid under light microscopy. Puchtler alkaline Congo-red stain has been the standard method of amyloid staining for a long time, but since this stain is relatively unstable and has low sensitivity, a control staining with positive specimens is absolutely essential.64 Daylon stain (also known as direct fast scarlet) is more sensitive: it is therefore of greater utility in detecting smaller amounts of amyloid deposition, but requires more careful observation because of a known tendency to overstain.64 Fluorescent microscopy is also useful for the diagnosis of amyloidosis. Thioflavin-S is a sensitive stain for amyloid deposition, and it has been used from long time along with Congo-red staining.65 Immunohistochemistry with fluorescent antibodies specific for precursor proteins is also a reliable diagnostic complement.
CAA severity grading
Two grading systems for CAA are commonly used in routine neuropathology. Olichney et al.66 proposed the scale: 0, no Aβ positive blood vessels; 1, scattered Aβ positivity in either leptomeningeal or intracortical blood vessels; 2, strong, circumferential Aβ positivity in either some leptomeningeal or intracortical blood vessels; 3, widespread, strong, circumferential Aβ positivity in leptomeningeal and intracortical blood vessels; 4, same as 3 with additional dyshoric changes. Vonsattel et al.67 graded CAA with respect to the severity of pathological changes in a given blood vessel: mild, amyloid is restricted to the tunica media without significant destruction of smooth muscle cells; moderate, the tunica media is replaced by amyloid and is thicker than normal; severe, extensive amyloid deposition with focal wall fragmentation or even double barreling of the vessel wall, microaneurysm formation, fibrinoid necrosis, and leakage of blood through the blood vessel wall.
Clinical features
CAA can be completely asymptomatic, especially since approximately 50% of individuals over 80 years of age display some pathology evidence of amyloid deposition as part of normal aging processes. However, amyloid deposition in cerebral blood vessels does favor development of several clinical conditions. Amyloid deposition can weaken cerebral blood vessels walls, causing rupture and therefore leading to both asymptomatic microbleeds and lobar ICH. Amyloid deposits can also obliterate the vessel lumen, leading to ischemia and related clinical manifestations (cerebral infarction, "incomplete infarction", leukoaraiosis). Focal neurological deficits, disturbances of consciousness, progressive cognitive decline, dementia, and death can occur as a consequence of these vascular mechanisms (albeit additional biological processes are also likely to be implicated).15
CAA-related ICH (CAA-ICH) accounts for 5-20% of all spontaneous (non-traumatic) ICH in elderly subjects. CAA-ICH tends to be lobar in location, due to the involvement of superficial cortical and leptomeningeal vessels, and often manifests as recurrent or multiple simultaneous bleeding events, because of the widespread nature of the angiopathy. Hypertension is less commonly associated with lobar hemorrhages than with non-lobar ICH.15 Increasing evidence is emerging that CAA may be a risk factor for thrombolysis-related intracerebral hemorrhage. CAAH and thrombolysis-related intracerebral hemorrhage share some clinical features, such as predisposition to lobar or superficial regions of the brain, multiple hemorrhages, increasing frequency with age, and an association with dementia.68 In vitro work showed that accumulation of amyloid-beta peptide causes degeneration of cells in the walls of blood vessels, affects vasoactivity, and improves proteolytic mechanisms, such as fibrinolysis, anticoagulation, and degradation of the extracellular matrix.68
Clinical diagnosis
While hemorrhagic stroke is the defining clinical characteristic raising concern for CAA, there are no pathognomonic clinical features of CAA-ICH. Headache, focal neurological deficits, seizures and altered level of consciousness occur in all ICH patients, based on hematoma size and location rather than pathophysiological mechanisms. Spontaneous bleeding due to CAA, as previously mentioned, can also be small and asymptomatic and is commonly referred to as a "microbleed".69,70 Microbleeds are part of the integrated clinical and imaging assessment leading to CAA diagnosis during life (see below), and have also been shown to correlate with risk of cognitive decline, functional dependence and lobar ICH recurrence.71 A recent study, based on assessment of microbleeds and leukoaraiosis in CAA-ICH and hypertensive ICH patients, suggested that both pathophysiological mechanisms might be present simultaneously in up to 25% of ICH patients.72
A definitive CAA diagnosis can only be formulated after histological investigation of affected brain tissue, obtained at autopsy or via brain biopsy. In practice evidence of CAA is very often found unexpectedly at post-mortem investigation. Non-invasive CAA diagnostic criteria have been therefore developed and refined in the past decade, in order to both improve and standardize diagnosis during life (Table 2).73 Additional refinement of these criteria based on inclusion of superficial siderosis among imaging markers of CAA has been recently proposed.74 Furthermore, positron emission tomography imaging with the beta-amyloid-binding compound Pittsburgh Compound B has been recently proposed as a potential noninvasive method for CAA detection in living subjects.75 A recent report suggested that severity of Pittsburgh Compound B retention is associated with risk of recombinant tissue-type plasminogen activator-associated intraparenchymal hemorrhage.76
Prevention and treatment
No evidence-based treatment or preventive strategy for CAA or CAA-ICH exists at this time. Corticosteroid treatment has been shown in some case reports and small series to ameliorate symptoms associated with CAA-related inflammation, possibly by reducing vasogenic edema.77 Other immunosuppressant treatments have been reported to influence the course of inflammatory CAA, but available evidence is extremely limited.78 However, recently reported results from the PROGRESS trial suggests that blood pressure control is likely to reduce risk of CAA-ICH recurrence.79 While CAA has been shown to represent a risk factor for thrombolysis-associated ICH and warfarin-related hemorrhagic stroke, no clear tools for risk prediction and stratification have yet been developed and validated. In particular, there is growing interest in the possible role of leukoaraiosis and microbleeds as surrogate markers for CAA severity, and therefore as possible tools for prediction of CAA-ICH.80 Of note, a recent study found an association between aspirin use and CAA-ICH recurrence, providing preliminary evidence of a possible association between microbleed burden and re-bleeding risk due to antiplatelet treatment.81 Similarly, in light of recent evidence associating statin treatment with increased risk for ICH recurrence additional research is necessary in order to determine the risks and benefits of lipid-loweirng treatment for CAA patients.82
Hereditary Cerebral Amyloid Angiopathy
Overall, hereditary forms of CAA (Table 1) are generally more severe than sporadic forms and often characterized by earlier age of onset, more severe clinical course, and earlier age of neurologic devastation and/or death.83-89 Unlike sporadic CAA, hereditary forms are exceedingly rare and tend to present in selected families in the form of autosomal dominant disorders. Both sporadic and hereditary CAA often cause cognitive impairment, but lobar ICH is not a consistent feature of all hereditary CAA forms. Specifically, hereditary CAA can be further classified in Aβ and non-Aβ forms, based on the accumulating peptide (or fragment). While involvement of leptomeningeal or cerebral vessels has been described in all familial syndromes, lobar ICH rarely dominates the clinical picture of non-Aβ CAA, with the remarkable exception of the non-Aβ Icelandic type. As for familial Aβ forms (Fig. 1), although some alterations in APP processing have been associated with corresponding mutations (particularly the Flemish mutation), their primary characteristic appears to be a modification of biochemical and cell biological properties of the peptide itself, including conformation, aggregation and fibril generation. Of note, APOE seems to play less of a significant role in hereditary than sporadic CAA, possibly reflecting the overriding role of the autosomal dominant Aβ mutation in determining Amyloid accumulation and therefore disease risk and clinical course.
Conclusion
CAA presents in both sporadic and hereditary familial forms. While hereditary forms are rare in the population and tend to affect younger individuals, sporadic CAA is a common disease of the elderly, its incidence and severity increasing with age. The emphasis placed on CAA in medical practice and research is justified by its association with cognitive decline, dementia and, more importantly, spontaneous lobar ICH. In the past decade a vast body of knowledge has been gathered on CAA pathogenesis, and efficient clinical and imaging tools have been developed to allow reliable diagnosis in life. However, further research efforts are required in order to identify targets for therapeutic and preventive interventions, aimed at limiting mortality, disability and neurological compromise associated with this CAA and CAA-related ICH.

 XML Download
XML Download