

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Human transmissible spongiform encephalopathy (TSE) includes classic Creutzfeldt-Jakob disease (CJD) and its variant form, Gerstmann-Sträussler-Scheinker syndrome (GSS), fatal familial insomnia, and kuru.1 Most cases of human TSE are sporadic, but it can also occur in genetic and acquired forms. GSS is a rare genetic form of TSE that was originally described in a large Austrian family, but it has never been reported in Korea.2,3 Typically, it is determined genetically by a range of mutations within the open reading frame of the prion protein gene (PRNP) on chromosome 20.4
The clinical spectrum of this syndrome is more diverse than that of sporadic CJD, including slowly progressive cerebellar ataxia and late cognitive decline. The disease progresses more slowly than dose sporadic CJD, and the overall disease duration may last several months to several years (median 5 years).2,3 The present report is of the first case of GSS (confirmed by PRNP analysis) in Korea. Distinctive MRI findings are also presented and discussed.
Case Report
A 46-year-old woman was admitted due to a slowly progressive gait and speech disturbance that had first appeared 3 years earlier. Initially, she experienced gait difficulty due to ataxia, which slowly progressed. Six months later, she noticed dysarthria. On examination at the previous hospital, her speech was explosive and gait was ataxic. She revealed an abnormal tandem gait, but the Romberg test was negative. The finger-to-nose test indicated slight dysmetria, but there was no dysdiadochokinesia. The deep tendon reflex response was normal and there were no pathological reflex responses. Motor and sensory function tests showed no abnormalities. Cognitive decline developed 1 year prior to admission to our hospital. Her neurological deficits had become increasingly aggravated and she had been bedridden for the previous 2 months. In the family history, two sisters out of seven siblings developed similar symptoms in their fourth and fifth decades, respectively, and both expired approximately 5 years after the onset of symptoms. Gene analysis for spinocerebellar ataxia (SCA1, 2, 3, 6, 7) was negative.
On examination, she appeared chronically ill. Her arousal level was good, but she lacked awareness. She could not cooperate during the neurological examination or respond to even simple commands due to severe dementia. She could not produce meaningful words, making only incomprehensible sounds due to language dysfunction and severe dysarthria. The only meaningful response was crying after a painful stimulus. Weakness in both of her lower extremities (MRC Grade III) along with signs of frontal lobe disease including the grasp, snout, palmomental, and glabellar reflexes were also evident. She fidgeted with her fingers and persistently exhibited perioral dyskinesia whilst mimicking chewing. She could not walk without assistance for 1 year prior to admission due to progressive ataxia. The deep tendon reflex response was decreased, but plantar responses were extensor bilaterally. Myoclonic movements were not observed.
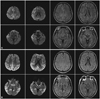
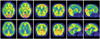
The results of blood studies - including chemistry, syphilis serology, human immunodeficiency virus serology, and a thyroid function test - were inconclusive. In brain MRI, high signal intensities over the entire hemispheric cortex were evident in fluid attenuated inversion recovery (FLAIR) imaging; this was significant in diffusion-weighted imaging (DWI). Follow-up images obtained 4 months later revealed the progression of the cerebral cortex signal changes from asymmetric to symmetric (Fig. 1). There was no definite change in the basal ganglia or cerebellum. Phosphorus-magnetic resonance spectroscopy (MRS) was performed using a 1.5-tesla system. FLAIR axial images and volumes of interest were obtained in the parietal cortex and basal ganglia. There were no significant abnormal MRS findings. 18F-FDG PET showed marked hypometabolism in the bilateral frontal and parietotemporal areas, with lesser amounts in the primary visual, motor and sensory cortices, basal ganglia, and cerebellum (Fig. 2). 14-3-3-protein was present in the CSF (Fig. 3). EEG performed during admission showed non-specific generalized slow waves, mainly in the theta range. PRNP analysis revealed a proline-to-leucine (P102L) mutation in codon 102 (Fig. 4). Additionally, a gene study revealed that two of her three children carried the same mutation. Her condition worsened, and she is currently being cared for at a nursing home.
Discussion
The original case of GSS was described in a large Austrian family with autosomal dominant inheritance. The typical clinical feature is slowly progressive cerebellar ataxia, beginning in the fifth or sixth decade (but onset may occur as early as 25 years), admixed with cognitive decline that appears at some time during the course of the illness.2,3 Unlike sporadic CJD, GSS involves a long period of illness: a mean of 5-6 years, ranging from 3 months to 13 years. Although the present case was a typical presentation with most of these classical features, several physicians failed to diagnose her case for 2 years, which is due to the rarity of the condition. Cerebellar symptoms were prominent even at an early stage, but there are several other diseases that present with ataxia. Several reports have indicated that MRI scans are normal during the early stage of GSS, with normal cerebellar findings being common in GSS patients.5 Therefore, this is useful for differentiating it from the cerebellar ataxia that usually appear as cerebellar atrophy and hypoperfusion in imaging studies. Although the present case exhibited obvious cerebellar symptoms, the cerebellum was normal in MRI and PET studies. Arata et al.5 suggested that the findings of dysesthesia and the absence of deep tendon reflexes at an early stage are important for differentiating between GSS and cerebellar ataxia.
Since the initial description of the P102L mutation which remains the most common, as confirmed in the descendants of the original Austrian family, various PRNP mutations have been described for GSS phenotypes.4 The onset occurred in the fourth decade for the younger sister of our case, which is less typical, but it is known that the clinical features vary for the P102L mutation.6 The death of her sisters after a neurological degenerative disorder with clinical similarities and the positive results of PRNP analyses for her children indicates that she had familial autosomal dominant GSS.
In general, the definitive diagnosis of a prion disease requires a brain biopsy or an autopsy with immunohistologic and genetic studies. Several ancillary tests are useful for this. EEG usually demonstrates generalized triphasic periodic sharp waves (PSW) in sporadic CJD. CSF testing for the 14-3-3 protein can also be helpful.7,8 However, these methods are not sensitive for genetic TSE. The present case tested positive
for CSF 14-3-3 protein but had non-specific EEG results. These findings are in agreement with previous reports; the EUROCJD collaborative surveillance project found that nearly 50% of GSS cases were positive for the 14-3-3 protein in CSF, while EEG tests showed typical pseudoperiodic activity in only 2 of 26 patients.9 Unlike the PSW or CSF 14-3-3 protein, MRI is not used in the World Health Organization CJD diagnostic criteria; however, its importance is increasing. MRI can be helpful for diagnosing specific types of human prion disease. For example, the pulvinar sign appears to be relatively specific to variant CJD. It was recently reported that DWI can assist with the early diagnosis and understanding of the relationship between lesions and neuropathology.10,11 One study showed that DWI is more sensitive for an early clinical diagnosis of CJD than are tests involving PSW, CSF 14-3-3 protein, and CSF neuron-specific enolase.10 Previous studies found that the usual neuro-imaging findings for GSS may be normal or show non-specific atrophy affecting the cerebral hemispheres and/or cerebellum and, rarely, decreased T2-weighted signal changes in the basal ganglia.12 Only a few studies have found abnormally high signal intensities in DWI of the cerebral cortex in GSS.13,14 The present case showed a high signal intensity in FLAIR imaging and DWI signals that were similar to those of sporadic CJD. Furthermore, the abnormally high DWI signal intensity was confined to the right hemisphere at 5 months prior to admission; it subsequently extended to both hemispheres, corresponding to disease progression. This case confirmed that DWI is more sensitive than conventional T2-weighted and FLAIR imaging in detecting GSS lesions, especially in the early stages. In patients with CJD, a high DWI signals corresponds to spongiform changes and astrocytic gliosis.11 Since the GSS syndrome is also included in the TSE category, a similar signal abnormality may be described by the same mechanism. Proton MRS has recently been reported to be useful in detecting subtle ongoing neural changes in patients with GSS syndrome.15 However, no abnormal finding in MRS was found in the present case. It is possible that our patient was in an early or intermediate stage in which neuronal damage was not severe.
This case is presented to alert clinicians that although it is a very rare human prion disease, GSS should be considered in the differential diagnosis when hereditary cerebellar ataxia and progressive cognitive decline develops. Unlike CJD, GSS is characterized by long periods of illness, and dementia develops late in the course of the illness. The utilization of diffusion-weighted MRI is suggested for the early diagnosis of GSS.


 XML Download
XML Download