

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cerebral small-vessel diseases (SVDs) are a major disease burden in most developed countries.1 SVDs usually manifest with lacunar infarction, intracerebral hemorrhage (ICH), and subcortical vascular dementia. Although hypertension and diabetes mellitus are known to be important risk factors for SVDs, many hereditary or idiopathic SVDs have also been identified. Among them, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common single-gene disorder of the cerebral small blood vessels and is caused by mutations in the Notch3 gene.2 The main clinical manifestations are recurrent stroke, cognitive deficits, migraines, and psychiatric disturbances. Although the exact prevalence of this disorder is not known, CADASIL has been reported worldwide in many ethnic groups. The increasing availability of diagnostic expertise and genetic testing has lead to a gradual increase in the number of reports about patients with CADASIL. This review summarizes the historical background, epidemiology, genetics, pathophysiology, characteristic clinical findings, neuroimaging, and treatment of this unique disorder.
History
CADASIL was possibly first reported in 1955, when Van Bogaert described two sisters with rapidly progressive subcortical encephalopathy of Binswanger's type.3 In 1977, Sourander and Walinder reported a Swedish family with multi-infarct dementia of autosomal dominant inheritance.4 They presented with pyramidal, bulbar, and cerebellar symptoms, a relapsing course, and gradually evolving severe dementia. The illnesses began in early adulthood (ranging from 29 to 38 years of age) and generally lasted for 10 to 15 years. Post-mortem studies showed multiple small cystic infarctions, localized in the central gray matter, white matter, and pons, caused by an occlusion of small intracerebral and leptomeningeal arteries. In the same year, Stevens et al.5 described familial vascular dementia in an English family with autosomal dominant inheritance. However, their mean age at the onset was 50 years (ranging from 39 to 57 years), and their mean age at death was 60 years (ranging from 56 to 65 years). The affected patients presented with a wide variety of transient motor, sensory, and other symptoms of vascular origin. The episodes varied considerably in frequency and severity within a family. The progressive phase of this illness was characterized by dementia, bulbar palsy with immobility, and urinary incontinence. Multiple small infarctions were seen in the basal ganglia, thalamus, and cerebral white matter at autopsy. Small arteries and arterioles showed medial swelling with vacuolar degeneration of smooth-muscle cells. A recent study demonstrated that hereditary multi-infarct dementia in the Swedish family reported by Sourander and Walinder was unrelated to CADASIL. This disorder does not affect the Notch3 gene, and these patients do not show characteristic features of CADASIL on pathologic examination.6 From the late 1980s to early 1990s there were several European reports of autosomal dominant cerebral SVDs characterized by recurrent stroke, progressive dementia, and migraine. In 1993, Tournier-Lasserve et al.7 applied linkage analysis to two large, unrelated French families presenting with CADASIL (they used the acronym CADASIL for the first time), and localized the disease to a locus on chromosome 19q12. Joutel et al.2,8 subsequently confirmed that the affected gene was Notch3 and identified several mutations therein.
Epidemiology
The exact prevalence of CADASIL is not known. According to a study from west Scotland, the disease prevalence was 1.98 per 100,000 adults with a probable CADASIL mutation prevalence of 4.14 per 100,000 adults.9 The researchers estimated the prevalence of CADASIL based on data from the regional registry. However, this figure could underestimate the true prevalence because CADASIL was frequently misdiagnosed at that time and they screened only four exons. Dong et al.10 determined the prevalence of CADASIL in a stroke population by screening exons 3, 4, 5, and 6 of the Notch3 gene in 218 subjects with lacunar stroke. Only one young patient exhibited a mutation in exon 4. The overall carrier frequency of the CADASIL mutation was 0.05%, and it was 2.0% and 11.1% for those with disease onset at ≤65 years and ≤50 years, respectively. CADASIL has been reported worldwide in many ethnic groups. The number of reported cases of CADASIL is gradually increasing as the clinical picture becomes more widely recognized and genetic testing becomes available. Most large reported clinical series have been from European countries including France, Germany, the UK, Finland, Italy, and the Netherlands.11-17 Reports from North America are relatively sparse, especially considering the high level of academic activity in this region. CADASIL families have been increasingly described in other world regions, including Asia, the Middle East, Australia, South America, and Africa.
Pathophysiology
The Notch3 gene encodes cell-surface receptors that transduce signals between neighboring cells.18 The Notch signaling pathway is highly conserved during evolution and is important for cell fate during embryonic development. Notch3 expression is restricted to vascular smooth-muscle cells (VSMC) in adults and participates in vascular development, differentiation, and remodeling.19,20 The NOTCH3 receptor is a single-pass transmembrane receptor of 2,321 amino acids. It consists of a large extracellular domain (ECD) with 34 tandem epidermal growth factor (EGF)-like repeats, three Notch/Lin12 repeats, a transmembrane domain, and an intracellular domain. Each EGF-like repeat contains six cysteine residues. The large ECD is noncovalently bound to the intracellular domain. Ligand binding leads to a proteolytic cleavage that translocates the intracellular domain to the nucleus. The intracellular domain activates the transcription factor RBPJk and other coactivators.
Knowledge of the effects of CADASIL-associated mutations on NOTCH3 function is important to understanding the molecular pathogenesis of this disorder. A small number of mutations located in the ligand-binding domain (EGF repeats 10 and 11) produce a significant reduction in transcriptional activity.21 However, greater numbers of mutations outside the ligand-binding domain lead to normal levels of signaling activity. Researchers have recently investigated the effects of mutations located in the ligand-binding domain in comparison with other common mutations.22
Patients with mutations in the ligand-binding domain showed significantly higher scores on cognitive scales, suggesting that reduced Notch3 signaling acts only as a modifier of the CADASIL phenotype. Therefore, Notch3 mutations do not cause functional abnormalities in the Notch canonical pathway, and loss of function of the Notch3 gene is probably not the cause of CADASIL.
Accumulation of the NOTCH3 ECD in the arterial wall is a characteristic finding of patients with CADASIL.19 A recent study showed that both wild-type and CADASIL-mutated NOTCH3 ECDs spontaneously form oligomers and higher order multimers in vitro.23 Notably, mutations causing CADASIL enhanced the formation of multimers in comparison with wild-type protein.
These findings implicate the neomorphic effects of CADASIL mutations as a pathogenetic mechanism underlying degeneration of VSMC. Nonetheless, whether the accumulation of the NOTCH3 ECD is the cause or the result of VSMC degeneration is not known. At present, the exact molecular mechanisms linking Notch3 mutations to degeneration of VSMC are uncertain.
Genetics
Over 170 different mutations have been identified in CADASIL, more than 95% of which are missense point mutations.24 Most of the mutations are located in exons 2-24, which encode the ECD of the NOTCH3 receptor. Of all the reported mutations, 62% are located in exons 3, 4, 5 and 8, and an additional 18% are located in exons 2, 6, 11, and 18. However, the reported distribution of mutations has varied with the world region.14,17,25 Almost all mutations lead to an odd number of cysteine residues in the affected EGF. However, a small number of reports describe mutations not involving cysteine.26-29 Because full exons were not screened26 or skin biopsies were not performed,27 whether cysteine-sparing mutations cause CADASIL remains debatable. Scheid et al.29 recently found deposits of granular osmiophilic material (GOM) between the smooth-muscle cells of small arteries and positive immunostaining with a NOTCH3 monoclonal antibody in patients with cysteine-sparing mutations. No other mutation was found in the remaining exons. However, a more complex rearrangement in the Notch3 gene that is not revealed by current molecular analysis remains a possibility. Two patients with de novo mutations have been described to date.30,31 These findings suggest that the absence of a family history does not rule out a diagnosis of CADASIL, and hence CADASIL may be more frequent than currently thought. In addition, patients homozygous for CADASIL mutations have been described, and their clinical features lie within the normal spectrum of CADASIL in comparison to age-matched heterozygous patients.32 Therefore, CADASIL follows the typical pattern of an autosomal dominant disease in which heterozygous and homozygous patients are clinically indistinguishable.
The founder effect refers to a type of genetic drift that accounts for elevated disease frequencies in small isolated populations. Several reports have suggested that CADASIL is influenced by this phenomenon. For example, evidence of a founder effect has been seen in both island populations, such as Taiwan and Jeju, and inland areas, including Finland and middle Italy, due to observed clustering of the same mutation.14,17,33,34 Finally, a novel heterozygous missense mutation (c.4544T>C) in exon 25 of Notch3 gene was reported in a patient with cerebral SVD but lacking deposits of GOM and NOTCH3 accumulation.35 This mutation results in increased canonical Notch3 signaling. This finding suggests that CADASIL is not the only cerebral SVD caused by Notch3 mutations.
Pathology
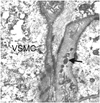
On autopsy, the brains of CADASIL patients show mild diffuse cerebral atrophy with multiple lacunar infarctions in the periventricular white matter, basal ganglia, thalamus, mesencephalon, and pons.36 The centrum semiovale shows diffuse loss of myelin with preservation of the subcortical U-fibers. Although on gross examination the cerebral cortex appears to be unaffected, neuronal apoptosis may be found in cortical layers, the severity of which correlates with the extent of ischemic lesions in the white matter.37 Histologically, medullary and leptomeningeal arterioles show thickening of the media. In addition, the walls of affected arterioles show degeneration of VSMC and deposition of granular eosinophilic material. Arteriolar stenosis is frequently observed along with severe thickening of arteriole walls that is more pronounced in the white matter than in the gray matter.38 Although the walls of lenticular nucleus arterioles were also thickened, definite stenosis was not observed in this region in comparison to the white matter.39 With the exception of the granular deposits, other pathologic findings did not differ significantly from those observed in other common cerebral SVDs. GOM refers to small or medium-size (20-40 µm) extracellular electron-dense granular deposits that are observed on electron microscopy (Fig. 1) and located near the VSMC of arterioles or veins.36,40 The exact composition of GOM is disputed. Joutel et al.19 showed that NOTCH3 ECDs accumulate at the cytoplasmic membrane of VSMC, in close vicinity to but not within the GOM. However, other researchers have used immunoelectron microscopy to show that NOTCH3 ECDs are abundantly and evenly distributed within the GOM, suggesting that they are a major component of GOM.41 Ultrastructural studies have shown destruction of VSMC and the presence of GOM in muscle and skin biopsies from patients with CADASIL. These findings have special clinical importance because skin and muscle biopsies could be useful for diagnosing CADASIL.42,43
Clinical Features
Ischemic stroke
Ischemic stroke is the most common clinical manifestation of CADASIL, being present in 60-84% of patients (Table 1).11,12,44 The mean age at stroke onset is reportedly 41-49 years and most of the patients present with typical lacunar syndrome manifesting as pure motor or pure sensory strokes, dysarthria or clumsy hand syndrome, and ataxic hemiparesis. Some patients show an insidious or stepwise evolution lasting up to several days. Although one study found that large cerebral arteries were involved in 20-30% of the patients, symptomatic stenosis was rare.45 ICH has been described only sporadically in patients with CADASIL. However, recent studies from Korea and Taiwan found that 25% of symptomatic patients with CADASIL had ICHs, with their development being closely related to the number of cerebral microbleeds (CMB).33,46 The presence of ICHs was found to be independently associated with poor clinical outcome in our patients with CADASIL (unpublished data).
Almost 70% of the patients who suffer from an initial stroke experience recurrent strokes, and some of them develop vascular parkinsonism and pseudobulbar palsy following recurrent subcortical infarctions.44 The mean incidence rate of stroke during a 2-year follow-up of 80 CADASIL patients was found to be 10.4 per 100 person-years.47 Vascular risk factors have been found only infrequently in patients with CADASIL in large clinical series, with hypertension reported in 6.6-28.6% of the patients and diabetes mellitus in 4.0-9.5% of the patients. The presence of vascular risk factors was not associated with age at the onset or presence of stroke in CADASIL patients.16
Cognitive deficits
Cognitive deficit is the second most frequent symptom, being found in about 60% of CADASIL patients. Dementia has been observed in 19-41% of the patients, with two-thirds of patients developing dementia by 65 years of age.11,12,16,44 However, recent studies from Asia found a relatively low (4.8-6.0%) prevalence of dementia in spite of the advanced mean age of the patients.33,46 However, because both studies found a dominant R544C mutation, this low prevalence of dementia might reflect an inherent characteristic of that mutation. Patients with CADASIL show characteristic frontal-lobe cognitive deficits, including problems with executive function, working memory, and verbal fluency. These symptoms can be confirmed by detailed neuropsychological testing even before the patient experiences an ischemic stroke.48,49 Patients with episodic memory dysfunction exhibit difficulties in retrieval rather than encoding. As the disease progresses, patients begin to show cognitive deficits typical of subcortical vascular dementia. The severity of cognitive deficit is closely associated with the number of lacunar infarctions seen in brain magnetic resonance imaging (MRI). However, volume of the white-matter high-signal-intensity (HSI) lesions was not significantly associated with the severity of the cognitive deficit in CADASIL after adjusting for age.50
Migraine
Migraine is the most common initial symptom in patients with CADASIL, with a reported incidence of 22-77%.11,12,16 Migraine usually starts around 20 years of age, and appears in 90% of patients by 40 years of age. Eighty percent of migraine patients report a preceding aura, and substantial numbers of patients experience basilar migraine, hemiplegic migraine, and migraine with prolonged aura. Compared with migraine in the general population, migraine in CADASIL patients has an older age at onset and higher incidence of aura, and it presents with atypical aura. The intensity and frequency of migraine attacks tend to decrease, with a small number of patients never developing migraine after they experience the first ischemic stroke. Nonetheless, the exact mechanism of migraine attack in patients with CADASIL is uncertain. Because migraines normally appear 10 years earlier than the ischemic stroke and the clinical features do not differ between patients with and without migraine, these headaches are probably not caused by brain ischemia. Interestingly, Asian studies, including those from Korea, Japan, and China, have found a lower prevalence (0-26.9%) of migraine, making the clinical detection of CADASIL even more difficult in these regions.26,33,34,51
Psychiatric disturbance
Psychiatric illness occurs in 20-41% of patients with CADASIL. Mood disorders are the most common,11,12,44,52 with the other psychiatric illnesses including conversion disorder, anxiety disorder, behavioral and personality disorder, psychosis and delusion, drug dependence, and alcoholism. Psychiatric disturbances in patients with CADASIL usually appear during the course of the disease, but they seldom represent the symptom at onset and are transient in most cases. Most psychotic events occur in patients with underlying dementia. Apathy is a frequent clinical feature of vascular dementia, and a recent study found that 41% of CADASIL patients had apathy apart from depression.53 Apathetic patients were older than nonapathetic subjects, had a lower cognitive score, had greater global disability, and presented with higher frequencies of depression and additional neuropsychiatric symptoms.
Other manifestations
Typically 5-10% of CADASIL patients develop epileptic seizures, which normally occur late in the course of the illness.11,12,16 Most patients that develop a seizure disorder have a history of stroke and dementia. Patients typically present more frequently with generalized tonic-clonic seizures than with focal seizure. A small number of patients present with acute reversible encephalopathy with fever, confusion, coma, and seizure lasting several days. A high prevalence (47%) of large right-to-left shunt was recently found in patients with CADASIL using a transcranial Doppler.54 However, the clinical significance of this presentation is uncertain because no clinical or MRI differences have been found between patients with and without shunt.
Clinical Course and Prognosis
The clinical course of CADASIL is highly variable even within families, with the age at the onset of stroke, dementia, and migraine possibly differing by 20 years among members of the same family.12,16 Therefore, there appears to be a weak correlation between genotype and phenotype. Some patients show a rapid progression to severe disability, whereas other patients remain stable or even improve. A large retrospective study from Germany found that conventional vascular risk factors such as hypertension, diabetes mellitus, and hypercholesterolemia did not influence the age at the onset or development of stroke.15 The median survival time of men in that study was significantly shorter than that expected from German life tables (64.6 vs. 69.3 years, p=0.01). In contrast, the median survival time of women was not significantly reduced (70.7 vs. 72.2 years). Although the median age at the onset of stroke did not differ between men and women, the median ages at the onset of the inability to walk without assistance, bedriddenness, and death were significantly lower in men than in women. Pneumonia was the most frequent cause of death, followed by sudden unexpected death and asphyxia.
Brain Imaging
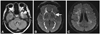
The most characteristic brain MRI findings in patients with CADASIL are white-matter HSI lesions and multiple lacunar infarctions (Fig. 2).13,55 On T2-weighted images, most patients exhibit HSI lesions in the white matter, more frequently in the periventricular and deep white matter than in the superficial white matter. HSI lesions start to appear in the subcortical white matter at 20 years of age in the absence of deep infarcts. These lesions then subsequently extend into the basal ganglia, thalamus, and brainstem. The involvement of the anterior temporal lobe and external capsule is characteristic, in contrast to sporadic ischemic HSI lesions. The sensitivity and specificity of the anterior temporal lobe lesions for the diagnosis of CADASIL were 90% and 100%, respectively.56 Unlike HSI lesions in other white-matter regions, those found in the anterior temporal areas were more severe even in younger subjects. The sensitivity and specificity of external capsule involvement were 93% and 45%, respectively. Lacunar infarctions usually appear by 30 years of age in the subcortical white matter, basal ganglia, thalamus, internal capsule, and brainstem. Lacunar infarcts were found in 75% of patients aged 30-40 years, and their number increased with age. CMB were found in 31-69% of patients and were located in various parts of the brain, preferential in the cortical/subcortical regions, white matter, thalamus, and brainstem. They were always found outside of ischemic lesions. Among the lesions observed in conventional MRI in CADASIL patients, the volume of lacunar lesion and the presence of CMB were independently associated with cognitive function and disability.57,58 Diffusion-tensor imaging detects ultrastructural tissue damage even in areas that appear normal in conventional MRI. The mean diffusivity was found to be the main predictor of clinical progression in patients with CADASIL.59 Recent studies have found that cerebral atrophy is independently associated with the extent of cognitive decline in patients with CADASIL, as with other common sporadic subcortical ischemic vascular diseases.60 The change in brain volume seems promising as an adjunct outcome measure in future interventional trials.
Brain MRI, single-photon-emission computed tomography (SPECT), and positron-emission tomography (PET) reveal that the cerebral blood flow (CBF) is lower in patients with CADASIL than in healthy controls. Hypoperfusion on brain SPECT is correlated with MRI ischemic lesions in demented patients showing very low blood flows.61 Chabriat et al.62 demonstrated significant reductions in both absolute and relative CBFs in the white matter of CADASIL patients using an MRI bolus-tracking method. This reduction was also more severe in demented than in nondemented patients. However, no significant change in absolute CBF was observed in the cortex of patients with CADASIL. PET showed that CBF in the cerebral white matter was lower in young CADASIL patients than in controls even before the patients had suffered a stroke.63
Diagnosis
Proper diagnosis of CADASIL is important for several reasons. First, the clinical course and prognosis differ between patients with CADASIL and those with other common cerebral SVDs. Second, proven therapies for ischemic stroke, including thrombolytics, antithrombotics, antihypertensive agents, and statins, have not been validated for patients with CADASIL. Clinical manifestations including migraine, recurrent subcortical stroke, psychiatric illness, and a family history of stroke, migraine, and dementia may be helpful in differentiating CADASIL from more common sporadic SVDs. One study showed that the long clinical course (several decades) and variable onset of symptoms even within families can result in as many as 35% of CADASIL patients being initially misdiagnosed with multiple sclerosis, dementia, or central nervous system vasculitis.64 At the time of first presentation, only 34% of CADASIL patients have first-degree relatives with a history of stroke, and only 16% have first-degree relatives with a history of stroke prior to 50 years of age. Therefore, an initially negative family history should not exclude considering CADASIL in daily routine clinical diagnoses.
The initial detection may be even more difficult in Asian populations for several reasons:26,33,46 1) Asians show a lower prevalence of migraine, 2) studies from Asian regions describe a lower involvement (22-43%) of the anterior temporal lobe in brain MRI, and 3) the high prevalence of vascular risk factors for SVD such as hypertension in patients with CADASIL increases the difficulty of differentially diagnosing CADASIL in Asian patients. Currently the cause of such differences between CADASIL patients from Asia and Europe is unclear. Regional differences in genotype alone may not explain this because 1) there are no significant differences in phenotypes among various genotypes in Asian CADASIL patients and 2) the age at the onset of stroke, dementia, and migraine differ among members of the same Asian families. Our recent analysis showed that the number of lacunar infarctions per patient and the volume of white-matter HSI lesions were both lower in Asian patients than in European patients in spite of the mean age being higher in the former (unpublished data). Asian patients with CADASIL seem to show a milder clinical course, although some of them develop ICH, which is associated with hypertension. The lower prevalence of ischemic stroke and lacunar infarction may explain the milder clinical course in Asian populations. The cause of such differences is unclear currently. The presence of CMB and history of hypertension were closely related to the development of ICH. Because ICH is the one of the most important factors in determining disability, the strict control of blood pressure may be a crucial management issue for Asian CADASIL patients. Currently there are certain limitations to differentiating CADASIL from sporadic SVD based on a clinical examination, family history, and neuroradiological findings. More epidemiologic work is required, especially in regions from where CADASIL reports are currently lacking.
Genetic analysis or pathological studies are used to confirm the diagnosis in a patient suspected of having CADASIL following clinical evaluation or brain MRI. Screening whole exons is time-consuming and expensive. Therefore, it is more efficient to initiate screening for mutations located in exons 3, 4, 5 and 8, which cover 62% of known mutations. In addition, including exons 2, 6, 11, and 18 increases the coverage to 80%.24 If the dominant mutations in that region are already known, a simple test using restriction enzyme digestion could be performed instead of more complicated DNA sequencing. A skin biopsy is easy to perform and its diagnostic specificity is very high since GOM has not been detected in any other disorders. However, the reported sensitivity has varied from 45% to 100%.65,66 A recent large retrospective study involving 131 Finnish CADASIL patients detected GOM in all 131 mutation-positive patients.24 Therefore, using electron microscopy to detect GOM in skin biopsies is a highly reliable diagnostic method. To maximize the diagnostic sensitivity in skin biopsies, researchers have suggested that the skin biopsy should be deep and large enough to include small arterioles located in the deep dermis or upper subcutaneous tissue. If the first biopsy results are negative, a repeated biopsy should be considered. Immunostaining for NOTCH3 ECDs increases both the sensitivity and specificity to more than 90%.67
Treatment
At present there is no specific treatment for patients with CADASIL. The primary treatments are genetic counseling, supportive care, and medications for treating depression, migraine, and the secondary prevention of ischemic stroke. Some studies have shown that acetazolamide is an effective treatment for migraine in patients with CADASIL.68,69 The safety and efficacy of migraine-preventing medications (e.g., beta-blockers and antiepileptic drugs) and migraine-aborting drugs (e.g., ergot derivatives and tryptans) have not been demonstrated. It is reasonable to control vascular risk factors such as hypertension and hypercholesterolemia considering the high recurrence rate of ischemic stroke in CADASIL. However, the effect of such treatments on the prognosis of CADASIL is uncertain. A two-center cohort study found that blood pressure and hemoglobin A1c were significantly associated with the presence of CMB, which in turn was independently associated with greater disability but not with dementia.58 Our previous study showed that the presence of CMB was associated with the development of ICH, which was significantly associated with poor functional outcome.46 These findings suggest that controlling the blood pressure and serum glucose may improve the clinical course of this disorder. Smoking should be avoided because it was found to be independently associated with being younger at the onset of stroke.16 However, the effects of antiplatelet agents and anticoagulants are uncertain. Anticoagulants are not recommended due to several reports of the development of hemorrhagic stroke. A recent randomized double-blind trial with the cholinesterase inhibitor donepezil showed no effect on the primary endpoint in CADASIL patients with cognitive impairment.70 Improvements were noted on several measures of executive function, but the clinical relevance of these findings was unclear. Conventional cerebral angiography resulted in neurological complications in up to 70% of patients in one study, and thus should be avoided in patients suspected of CADASIL.71
Conclusion
CADASIL is the most common hereditary cerebral SVD, and the number of reports on patients with CADASIL is steadily increasing. Although specific treatments for CADASIL patients are currently unavailable, the disease-causing mutations are well described and active research into the pathophysiology of the disease is underway. New therapies that could prevent or slow the degeneration of VSMC will probably emerge in the near future, as will clinical studies using specific neuroimaging surrogate markers to monitor the treatment course. CADASIL is a prototype single-gene disorder that has evolved as a unique model for studying the mechanisms underlying cerebral SVDs. At present, the incidence and prevalence CADASIL seem to be underestimated due to limitations in clinical, neuroradiological, and genetic diagnoses of this disorder.

 XML Download
XML Download