

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Pantothenate-kinase-associated neurodegeneration (PKAN), formerly called Hallervorden-Spatz syndrome, is an autosomal recessive neurodegenerative disorder characterized by progressive extrapyramidal signs, visual loss, and cognitive impairment.1 PKAN is caused by mutations in PANK2, which is a gene located on chromosome 20p13 and encodes pantothenate kinase, the key regulatory enzyme in coenzyme-A biosynthesis.2 PKAN belongs to a group of neurodegenerative diseases that includes neurodegeneration with brain iron accumulation (NBIA), infantile neuroaxonal dystrophy, aceruloplasminemia, and neuroferritinopathy. Iron accumulation in the globus pallidus can be frequently detected by MRI in a characteristic pattern referred to as the "eye-of-the-tiger" sign.3
Two main phenotypes of PKAN have been reported: classical and atypical.1 Patients with classical PKAN typically present in the first decade of life with severe extrapyramidal signs, progressing relentlessly and leading to independent walking disability within 10-15 years after onset. However, atypical PKAN cases show later onset, less severe extrapyramidal signs, and slower progression. Here we describe a patient with atypical PKAN presenting with adult-onset, slowly progressive parkinsonism due to a novel PANK2 mutation.
Case Report
A 45-year-old man was admitted to our hospital because of a 10-year history of tremor involving both hands. His past medical history was unremarkable. There was no history of perinatal complications and he met normal developmental milestones. From the age of 35 years, the patient began experiencing bilateral postural hand tremor. He also felt a mild slowness of speech. Symptoms worsened gradually over several years. At the age of 40 years he was unable to continue working as a hairdresser due to the hand tremors. He had three brothers and two sisters; the oldest brother also had mild hand tremors that did not prevent him performing the activities of daily living independently.
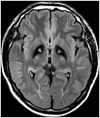
Upon neurological examination, the patient had a masked face and spoke in a monotonous voice. He presented with mild bradykinesia and rigidity involving all of his extremities. Horizontal saccadic eye movements were slow and fragmented. Pursuit eye movements were unremarkable. Outstretching his arm elicited a postural hand tremor with a moderate amplitude and a frequency of 5-6 Hz. Tongue tremor, which seemed to be grossly synchronous with hand tremor, was also observed. On walking, his gait velocity was slow with decreased arm swing on his right side. There were no motor or sensory abnormalities. Deep-tendon reflexes were normoactive and without the Babinski sign. Complete blood count, biochemical screening, erythrocyte sedimentation rate, C-reactive protein, and urine were normal. Serum ceruloplasmin and copper concentrations were normal. The serologic test for human immunodeficiency virus and assays for anti-nuclear antibodies were negative. Brain MRI revealed bilateral and symmetric hypointensity with medial hyperintensity in the globus pallidus on fluid attenuation inversion recovery imaging. This feature was in accordance with the "eye-of-the-tiger" sign (Fig. 1).
A mutation analysis (Fig. 2), in which PCR sequencing was utilized to identify PANK2 mutations, revealed three mutations: two in exon 3 (Asp378Gly and Leu385CysfsX13) and one in exon 4 (Arg440Pro) of the gene. An A-to-G change at nucleotide 1133 results in the substitution of aspartate for glycine at amino acid 378 (D378G), an insertion of threonine at nucleotides 1154-1155 results in a frameshift at amino acid 385 (L385fs), and a G-to-C change at nucleotide 1319 results in the substitution of arginine for proline (R440P). Other members of his family did not consent to genetic testing. There was no overt response of the patient's parkinsonism to the introduction of L-dopa. However, his hand tremor improved partially after treatment with pramipexole dihydrochloride (0.5 mg three times daily) and clonazepam (0.25 mg twice daily).
Discussion
Phenotypically, the present case is compatible with atypical PKAN in terms of adult onset, its slowly progressive nature, and its parkinsonian presentation. Several cases of NBIA that presented with adult-onset parkinsonism have been reported in the literature, and in their series of patients with NBIA, Thomas et al.4 reported that four out of ten patients with a PANK2 mutation initially presented with parkinsonism. Regarding the phenomenology of PKAN-associated movement disorders, patients with an early onset predominantly presented with dystonia, whereas adult-onset patients exhibited parkinsonism as the initial symptom. Although parkinsonism has frequently been observed in early-onset PKAN patients, other neurologic abnormalities of dystonia, pyramidal signs, and cognitive impairment were generally combined.5,6 Accordingly, pure parkinsonism is not an unusual presenting symptom of atypical PKAN.4
The genetic analysis revealed two PANK2 mutations in exon 3 (Asp378Gly and Leu385CysfsX13) and one in exon 4 (Arg440Pro). Of those, D378G is an already established mutation found in PKAN.7 However, L385fs and R440P are novel mutations that have not been reported previously. Insertion of threonine at nucleotides 1154-1155 in exon 3 results in a frameshift mutation with a premature termination of translation at amino acid 385. It may be a null allele that causes a loss of function. A novel substitution at nucleotide 1319 in exon 4 replaces arginine in position 440 with proline. Arginine provides a positive hydrophilic charge with basic side chains, while proline is a cyclic amino acid that has a neutral hydrophobic charge. Therefore, it may be speculated that the arginine-to-proline substitution results in loss of the functional properties of the PANK2 protein.
Regarding the association between PANK2 mutation and clinical phenotype, two loss-of-function alleles and residual enzymatic activity of PANK2 are known to significantly determine the age of onset of the disease, but have a much weaker influence on the rate of disease progression.6 Similarly, Thomas et al.4 suggested that patients with atypical PKAN were more likely to have mutations resulting in amino- acid changes. Although we were unable to determine the enzymatic activity of PANK2 in our case, this compound heterozygous mutation may have been responsible for the adult-onset and delayed progressive nature of the disease.
Mutations on both alleles of PANK2 are identified in about 50% of patients with clinical features of PKAN, and in more than 98% of patients with radiographic features of the disease.7 As mentioned previously, parkinsonism is not an unusual presenting symptom in patients with atypical PKAN, and it is important for physicians to consider PKAN in the differential diagnosis of patients presenting with early-onset parkinsonism.

 XML Download
XML Download