

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The skeletal muscle sodium channel comprises a principal pore-forming and voltage-sensing subunit (the alpha subunit), which is associated with an accessory beta-1 subunit. Its alpha subunit is encoded by the gene SCN4A, which is located on chromosome 17q23-25,1 comprises 24 exons with a 5.5-kb open reading frame,2 and is associated with various neuromuscular disorders. The beta-1 subunit has not been reported to be linked to any human disease.
More than 40 different types of SCN4A mutation have been identified, most of which are single-base substitutions producing missense mutations.3 In contrast to the chloride and calcium channelopathies, which produce relatively uniform phenotypes, SCN4A mutations produce several clinically distinct skeletal muscle disorders including hyperkalemic periodic paralysis (HYPP), paramyotonia congenita (PMC), potassium-aggravated myotonia (PAM), hypokalemic periodic paralysis, and congenital myasthenic syndrome.4-8 Previous studies have also shown that certain SCN4A mutations are associated with a specific phenotype, and it has been suggested that the mutation-specific differences in aberrant channel gating behavior underlies the genotype-phenotype correlation in skeletal muscle sodium channelopathy.9
In this context, we studied six Korean patients with SCN4A mutations who showed various clinical types of nondystrophic myotonia and periodic paralysis syndromes. We reasoned that this would help to clarify the genotype-phenotype correlations in Korean patients with this group of disorders. We therefore performed a mutational analysis of SCN4A in these patients, and analyzed their clinical features in detail.
Methods
Subjects
Six unrelated Korean patients with nondystrophic myotonia and periodic paralysis syndrome were included in the study. The patients comprised one patient with HYPP, one with PMC, and four with pure myotonia. Mutation of CLCN1 had previously been excluded in all four patients with the pure myotonia phenotype, by full sequence analysis of its coding regions.10 The control group comprised 100 healthy Koreans. All of the patients and controls provided written informed consent to participate in this study, which was reviewed and approved by the Pusan National University Hospital Institutional Review Board.
Clinical evaluation
A detailed clinical history was taken from all patients, and all underwent a standard neurological examination. History-taking revealed several clinical data, including age of onset, first clinical symptom, the most disabling symptom at the time of presentation, and family history. On neurological examination, special notice was taken of the presence of muscle atrophy or hypertrophy, distribution of muscle weakness (if any), status of the deep-tendon reflexes, and types of maneuver that provoke myotonia. All patients submitted to electrodiagnostic studies, including routine nerve conduction studies and needle electromyography (EMG) from at least two different muscles in each extremity. Routine laboratory tests were performed, including complete blood count, liver and renal function tests, thyroid function tests, blood glucose, electrolytes, serum creatine kinase levels, chest X-ray, and electrocardiogram. Muscle biopsy procedures were performed in three patients. The biopsied muscle samples were flash frozen in isopentane prechilled with liquid nitrogen and then processed for routine histochemical reactions including hematoxylin-eosin, modified Gomori trichrome, NADH, and ATPase stains.
DNA analysis
The SCN4A mutations were identified by direct sequence analysis of whole coding regions of the gene. DNA was extracted from the patients' anticoagulated whole blood, and 21 sequence specific primer pairs covering the entire coding region of SCN4A were subjected to PCR amplification. Primer sequence data and information on the PCR conditions are available upon request.
The amplified PCR products were separated on 2% agarose gels, purified, cycle-sequenced with PCR primers using the BigDyeTerminator Sequencing Kit (PE Applied Biosystems, Foster, CA, USA), and electrophoresed using an ABI PRISM 3730XL DNA analyzer (PE Applied Biosystems).
PCR-restriction fragment length polymorphism (RFLP) was conducted on patient 3 and the 100 normal controls to confirm the presence of the novel c.673C>T (p.R225W) mutation found in this patient and to establish that it is not a benign polymorphism. The mutation was identified using the restriction enzyme Aci I to discriminate the mutated allele from normal alleles. The primer pair 5'-TGCACTGTCCT TCCCAACCC-3' (forward) and 5'-GCCTCTCAAACGCC CATCCT-3' (reverse) was used for the PCR reaction. Since the mutated allele will lose the normal Aci I restriction site, a heterozygous mutation of c.673C>T will produce three DNA bands at 472, 245, and 227 bp, while normal alleles will produce bands at only 245 and 227 bp. All enzyme digestion reactions were conducted at 37℃ for 2 hours, and the electrophoresis was performed on 2% agarose gel.
Results
SCN4A mutations
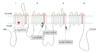
The sequence analysis revealed four different mutations including one that has not been described previously. The types and locations of the identified mutations are summarized in Figs. 1 and 2, and Table 1. A novel missense mutation, c.673C>T, was identified in exon 5 from patient 3, which is expected to change a codon for arginine into tryptophan at position 225 (p.R225W). This is considered a pathogenic mutation because 1) we were unable to observe the same alteration in any of the 100 normal controls using PCR-RFLP analysis (Fig. 3A), and 2) the mutated amino acid arginine at position 225 is highly conserved between different species in multiple alignment analysis of the amino acid sequence (Fig. 3B).
Clinical features
The clinical features of the six patients are summarized in Table 2. They comprised six men with ages of onset from birth to 30 years. The four patients with the pure myotonia phenotype (patients 1, 2, 3, and 6) had myotonia with the warm-up phenomenon, which initially affected predominantly the lower extremities. In this group of patients, muscle stiffness was provoked by voluntary contraction of the skeletal muscles after a period of rest, and typical myotonic discharges were observed on EMG. Patient 4 presented with myotonia that worsened by repetitive exercise and cold exposure, and was classified as having the PMC phenotype. EMG in this patient also revealed prominent myotonic discharges. Periodic weakness was the main symptom in patient 5, together with elevated serum potassium levels. Needle EMG also revealed myotonic discharges.
Discussion
We have identified four different SCN4A mutations (one of which has not been reported previously) in six unrelated Korean patients with nondystrophic myotonia and periodic paralysis syndrome. A novel heterozygous missense mutation c.673C>T (p.R225W) was found in patient 3, who presented with mild nonpainful pure myotonia (Table 1 and 2). This mutation is located at the cytoplasmic side of transmembrane S3 segment of domain I (DI/S3; Figs. 1 and 2), and is noteworthy because most of the previously reported SCN4A mutations are clustered in domains III or IV of the protein. Furthermore, it is the most proximal mutation ever reported in SCN4A. Thus far only two mutations affecting domain I have been reported.11,12 This mutation is also remarkable because there is only one previous report of an SCN4A mutation affecting the S3 segment (p.L1433R).13
Another mutation, c.2078T>C (p.I693T), was identified in patient 4, who exhibited a typical PMC phenotype. This mutation is located at the cytoplasmic link between S4 and S5 of DII, and has been reported previously in patients with PMC (Table 1, Figs. 1 and 2).14
The c.3466G>A (p.A1156T) mutation was shared by two of our patients who exhibited completely different phenotypes (Table 1, Figs. 1 and 2): while patient 2 had pure myotonia affecting the lower extremities, with warm-up phenomenon, patient 5 manifested as HYPP without clinical history or signs of myotonia. Although the mutation p.A1156T has been described previously both in patients with HYPP and PMC,15 it has never been associated with the pure myotonia phenotype, as in our patient 2. This phenotypic variability observed in individuals harboring the same SCN4A mutation strongly suggests that the genetic background-and perhaps other epigenetic factors-influences the clinical expression of particular mutations.
The c.3917G>A (p.G1306E) mutation, located at the cytoplasmic linker between domains III and IV (ID3-4), was also found in two patients, in both of whom it manifested as pure myotonia (patients 1 and 6). This mutation had been reported in patients with PAM and has been functionally characterized (Table 1, Figs. 1 and 2).16,17
Our study shows that SCN4A mutations produce different phenotypes of nondystrophic myotonia and periodic paralysis syndrome. We have also shown that certain SCN4A mutations may not be associated with specific clinical phenotypes, suggesting poor genotype-phenotype correlations in skeletal muscle sodium channelopathy.
In sodium channelopathy, there is a group of patients who manifest with pure myotonia. These patients exhibit one of several clinical phenotypes including myotonia fluctuans, myotonia permanens, and acetazolamide-responsive myotonia.18 They are distinct from those with PMC by the absence of paradoxical myotonia and cold sensitivity. Since many such patients have myotonia that becomes much more pronounced after the ingestion of potassium-rich food, the condition has been collectively called PAM. However, some patients with SCN4A-related pure myotonia do not exhibit such potassium sensitivity,11,19 and thus the condition may be more appropriately labeled sodium-channel myotonia. The myotonia in this group of patients is very similar to that observed in those with myotonia congenita (MC), and clinical differentiation may not be possible. The four patients with pure myotonia in our study had also initially been considered as having MC, but they were finally shown to have SCN4A mutations. A recent study involving the screening of SCN4A and CLCN1 mutations in large numbers of patients with nondystrophic myotonia found that 20% of patients who had been clinically diagnosed as MC had SCN4A mutations.20
Our study also showed that sodium channelopathy comprises highly variable clinical phenotypes, and that the genotype-phenotype correlation is not unequivocal. Although careful clinical evaluation is of great help, we suggest that a definite molecular diagnosis using sequence analysis of both SCN4A and CLCN1 is essential for the differential diagnosis of sodium channelopathy from other conditions, especially when a patient presents with pure myotonia.




 XML Download
XML Download