

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Erdheim-Chester disease (ECD) is a rare, sporadic, non-Langerhans form of histiocytosis of unknown etiology that may present with focal or diffuse manifestation. 1 It can involve various organ systems including the musculoskeletal, cardiac, pulmonary, gastrointestinal, and central nervous system. The most frequent systemic manifestations are bone lesions that are characterized by bilateral symmetric sclerosis of the diaphyseal and metaphyseal regions of the long bones.
Progressive cerebellar syndrome displaying cerebellar dysmetria, ataxia, dysarthria, and nystagmus is a rare neurological manifestation of ECD. In the published literature, there were only seven reported patients of cerebral ECD with progressive cerebellar syndrome, and in these patients, brain MRI generally reveals hyperintensity in both dentate nuclei without cerebellar atrophy. Here, we present the case of a cerebral ECD patient with progressive cerebellar syndrome in which the brain MRI showed cerebellar atrophy with focal lesions in the pons, middle cerebellar peduncle, and cerebellum.
CASE REPORT
A 37-year-old woman was admitted to the hospital because of multiple painful masses at the anterior aspect of both axillae, which she first detected 4 years ago. She is the mother of two normal children with no significant family history. Her medical history included type 2 diabetes mellitus of 4 years duration. She underwent incision and drainage of both axillae as the lesions were not amenable to simple aspiration. Histopathologic examination revealed foamy lipid-laden, non-Langerhans-cell histiocytes that lack grooved nuclei and were surrounded by medullary fibrosis. A few inflammatory cells, lymphocytes, plasma cells, and Touton-type giant cells were also seen (Fig. 1-A), and immunohistochemical stains were positive for CD68 and negative for S-100 protein (Fig. 1-B).
On the fourth day of her admission, the patient developed gait and limb incoordination with slurring of speech and was referred to our department. She also complained of generalized arthralgia, predominantly of the larger joints of her legs and arms.
On physical examination, she was found to have slightly raised, irregular, tender upper tibial shafts, and on neurological examination, she exhibited dysarthria with scanning speech, mild ocular overshoot, and moderate limb ataxia without tremor on finger-to-nose testing. She further displayed impaired smooth pursuit eye movement and gaze-evoked nystagmus. Muscle tone was normal and deep tendon reflexes of all four limbs were symmetric and hypoactive. Her gait was markedly ataxic.
Laboratory tests revealed an elevated CRP of 0.428 mg/dl (normal: <0.375 mg/dl) and ESR of 87 mm/h (normal: <9 mm/h). The serum vitamin E concentration was within normal limits. Thyroid, renal, and hepatic functions were normal. Cholesterol, triglycerides, ANA, and anti-DNA antibody levels were normal. Thoracoabdominal CT scan showed no abnormality of the kidney and aorta.
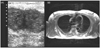
Axillary ultrasonography demonstrated multiple, bilateral subcutaneous abscess-like masses with internal echogenic content (Fig. 2-A), and an MRI of the axillae showed bilateral large ill-defined subcutaneous soft tissue masses (Fig. 2-B).
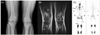
Plain film radiography demonstrated the typical lesions of ECD with patchy sclerosis and osteolysis in both distal femurs and proximal tibias (Fig. 3-A), and MRI revealed abnormal signal intensities of geographic pattern throughout the marrow of the diaphyses, metaphyses, and epiphyses in the same locations (Fig. 3-B). Whole-body bone scan showed abnormal tracer activity corresponding to the lesions of the femur and tibia and an additional area on the right humerus (Fig. 3-C).
Imaging of the brain with MRI in various sections exhibited the following: T1-weighted axial MRI showed symmetrical hyperintense signals in the tegmentum pontis, right basis pontis, and right temporal lobe (Fig. 4-A); Gadolinium enhanced T1-weighted coronal MRI showed enhanced lesions in right parietal lobe and middle cerebellar peduncle (Fig. 4-B); Gadolinium enhanced T1-weighted sagittal MRI showed moderate cerebellar atrophy and enhanced lesion in vermis part of the anterior lobe (Fig. 4-C); Gadolinium enhanced T1-weighted axial MRI showed an enhanced lesion in the right temporal lobe (Fig. 4-D). The clinicopathologic and radiological findings of our patient were sufficiently characteristic to be diagnostic of ECD.
Despite treatment with corticosteroids for 4 weeks, the patient's symptoms of cerebellar ataxia, dysmetria of the arms, and dysarthria did not improve. The symptoms slowly progressed over a follow-up period of 3 years to the point where she could no longer perform basic activities of daily living.
DISCUSSION
ECD is an extremely rare disease of unknown etiology, with distinct clinicopathologic and radiological findings in the absence of detectable serum lipid abnormalities.2 The clinical manifestations of ECD are not well defined, but men and women in the fifth through seventh decades of life appear to be affected. Pediatric cases are extremely rare.
Clinically, ECD ranges from a focal asymptomatic process to a multi-systemic fatal condition. Systemic complaints of fever, weight loss, and malaise are often present. Clinical manifestations are the result of histiocytic infiltration of various tissues. Bone pain is the most common presenting symptom of ECD, occurring in approximately half of patients, and is mostly located in the lower limbs.1
Skeletal involvement is the cardinal feature of ECD. Most commonly the long bones of the upper and lower extremities are involved bilaterally and symmetrically in ECD. Diffuse or patchy increased density, coarsened trabecular pattern, medullary sclerosis, and cortical thickening usually affect the diaphyses and metaphyses.1,3 The epiphyses are rarely involved.3 Our case demonstrated the usual symmetric osteosclerosis of the diaphyses and metaphyses of the long bones but also displayed an atypical radiographic findings of ECD, namely the involvement of the epiphyses.
Approximately half of ECD patients have extra skeletal manifestations. In a review of the literature, mass formation of ECD has been reported in various tissue sites such as retroperitoneum, neck, orbit, and breast, and demonstrate the same histopathologic findings as those of the bone marrow.3-5 Our case is again unique in that the same pathologic process presented itself as multiple masses in both axillae.
Because of nonspecific clinical presentations, ECD is often misdiagnosed. Histopathology plays a crucial role in the diagnosis of ECD, especially in differentiating it from other histiocytosis. It is well known that the majority of histiocytes in ECD are negative for S-100 protein and positive for CD68 in immunohistochemical staining.
Nerurological involvement is found in less than 30% of ECD patients and always associated with extra-neurological involvement.6 Skeletal involvement was the most common, followed by retroperitoneal, orbital, cutaneous, and cardiovascular involvement. In two-thirds of the cases, ECD began with extra-neurological manifestations, especially diabetes insipidus (DI). DI was known in 29% of total ECD and in 47% of ECD with neurological involvement.
Neurological manifestations of ECD are variable. Cerebellar and pyramidal syndromes are the most frequent and estimated to be 41% and 45%, respectively. Seizures, headaches, neuropsychiatric or cognitive problems, or cranial nerve palsies are also reported. Neurological disorders due to ECD occur in the course of the illness and are most often associated with intracranial tissue infiltration by foamy histiocytes.1,7,10,14
Among the neurological manifestations of ECD, progressive cerebellar syndrome is a rare finding. Recently, Weidauer et al.8 reported a 44-year-old man with cerebellar syndrome who developed slowly progressive cerebellar dysfunction (with a follow up of 24 months) displaying cerebellar dysmetria, ataxia, dysarthria, and nystagmus. Axial T2-weighted MRI images of the brain showed hyperintensity in both dentate nuclei without cerebellar atrophy. This same type of neurological involvement of ECD with cerebellar sparing has been reported in only seven other patients.9-15 Our patient also had ECD with progressive cerebellar syndrome, but the brain MRI findings were unique in that it demonstrated moderate cerebellar atrophy with focal lesions in the pons, middle cerebellar peduncle, and cerebellum. Although the pathogenesis of cerebellar atrophy is not clear, we suggest that lesions of the pons, middle cerebellar peduncle and cerebellum may have secondarily produced the cerebellar atrophy (pseudoprogressive olivopontocerebellar degeneration).
Treatment options include corticosteroids, radiotherapy, chemotherapy, and immunotherapy or combination therapy. None are highly effective and the disease is typically relentless in its course. There is no certain consensus on the treatment of ECD.1,7 There are only a handful of cases of ECD reported in the literature and the exact prognosis is still unknown. The prognosis for ECD may be related to the extent of visceral involvement. The average time of survival after diagnosis is 32 months. The most common causes of death include respiratory failure, pulmonary fibrosis, and heart failure. Brain or spinal cord involvement portends a very poor prognosis.
We report a rare case of cerebral ECD with progressive cerebellar syndrome in which, unlike previously published reports, the brain MRI shows cerebellar atrophy. In addition, clinicopathologic manifestations involving such locations as the axillae presenting as bilateral axillary masses and the epiphyses of lower extremity long bones are distinctly unusual.


 XML Download
XML Download