

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Charcot-Marie-Tooth (CMT) disease type 1A (CMT1A), the most common type of CMT neuropathy, is associated with duplication of chromosome 17p11.2-p12, whereas hereditary neuropathy with liability to pressure palsies (HNPP), which is an autosomal dominant neuropathy showing characteristics of recurrent pressure palsies, is associated with 17p11.2-p12 deletion. The proximal and distal CMT1A-REP repeats on 17p11.2-p12 are involved in unequal crossover resulting in CMT1A duplication and HNPP deletion.2 A gene dosage effect of peripheral myelin protein 22 (PMP22) is considered to be the chief mechanism underlying the development of CMT1A and HNPP.3 The genetic mechanism causing the CMT1A duplication is unequal nonsister chromatid exchange during spermatogenesis.4 Therefore, it has been reported that the prevalence of de novo duplications in CMT1A is much higher for those of paternal origin than for those of maternal origin.5
Even though CMT1A and HNPP are associated with the same locus, there have been no reports of the occurrence of these two mutations in a single family. Here, we report a de novo CMT1A patient of paternal origin in a family with HNPP harboring a deletion.
CASE REPORT
Patient 1 (proband, III-1 Fig. 1) was an 11-year-old girl with a normal birth. She had experienced gait abnormalities from infancy. At the age of 2 years, she was observed to walk ploddingly with frequent stumbling. At 5 years, a wasting of bilateral leg muscles was evident, and she wore a foot brace. The motor weakness and wasting were more severe in the lower distal extremities than in the upper extremities. Muscular stretch reflexes were absent, and vibratory sense and pain appreciation were impaired distally in both upper and lower limbs.
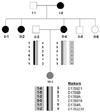
Patient 2 (proband's mother, II-4) was a 45-year-old woman born after a normal delivery. All early developmental milestones were achieved. At the age of 27 years, she experienced weakness of both upper extremities that lasted for 3 months after giving birth. She experienced sensory impairment of the right upper extremity after working hard. Also, her mother (I-2) and two sisters (II-1 and -2) experienced similar recurrent episodes of weakness and sensory impairments after the application of minor pressure. A neurological examination did not reveal any muscular atrophy. Muscular stretch reflexes were present and all sensory modalities were intact.
Nerve conduction was investigated in this family (Table 1). The proband showed markedly reduced motor nerve conduction velocities (NCVs) with prolonged distal motor latencies. However, patient 2 exhibited focal neuropathy with common entrapment sites. The median motor NCVs of the proband and her mother were 15.4 and 52.3 m/s, respectively (normal value in our laboratory ≥ 50.5 m/s). Stimulation at the wrist, elbow, and above the elbow evoked motor compound muscle action potentials with normal amplitudes, and no conduction block was observed in either patient (Fig. 2).
Mutational analysis of the family was performed (Fig. 1). Six microsatellite markers within 17p11.2-p12 (D17S921, D1S9B, D17S9A, D17S918, D17S4A, and D17S2230) were genotyped to determine the presence of duplication or deletion. We identified a 17p11.2-p12 deletion in patient 2 (II-4), and diagnosed her as HNPP. However, the proband (III-1) was determined as having a de novo duplication of paternal origin. The duplication contained both paternal haplotypes, indicating that the de novo mutation was caused by an unequal nonsister chromatid crossover at paternal meiosis. No other causative mutation in PMP22, MPZ, GJB1, EGR2, NEFL, PRX, or MFN2 was identified in this family.
DISCUSSION
CMT disease and HNPP are genetically and phenotypically heterogeneous peripheral neuropathies.1 CMT1A duplication and HNPP deletion are due to a reciprocal unequal crossover event between flanking CMT1A-REP repeats and the PMP22 gene locates within the 17p11.2-p12 region.2 There are several lines of evidence that alterations in the gene dosage of PMP22 are the main cause of the CMT1A phenotype.3 Namely, patients carrying one extra copy of PMP22 develop CMT1A, while patients with HNPP deletion have only one copy of PMP22.3 This is reflected in reduced mRNA and protein levels in sural nerve biopsy samples from HNPP patients.6 Myelin plays an important role in the saltatory impulse transmission along neuronal extensions and communication between neurons, and the level of PMP22 expression must be maintained within the critical range for proper peripheral nerve function.7
It is believed that CMT1A and HNPP are both demyelinating neuropathies, but their clinical, electrophysiological, and histopathological features are quite distinct.6 CMT1A patients with duplication develop slowly progressive sensorimotor length-dependent neuropathies associated with uniformly slow NCVs, and HNPP is characterized by recurrent episodes of focal entrapment neuropathies occurring as a result of trivial trauma or pressure.8 However, most HNPP patients exhibit conduction slowing only at sites of mechanical compression, but mild overlap of clinical features with CMT may lead patients with HNPP to be misdiagnosed as having CMT1A.6 Moreover, all heterozygous patients with a PMP22 point mutation (T118M) had clinical and electrophysiological features of a neuropathy similar to HNPP by a partial loss of PMP22 function.7
CMT is frequently classified into type 1 (the demyelinating form; CMT1) and type 2 (the axonal form; CMT2).1 The primary defect in CMT2 patients is neuronal, with them exhibiting slightly reduced (>38 or even normal NCVs.9 Recent studies have defined intermediate-type CMT, which has electrophysiological and pathological features of both CMT1 and CMT2.10 Before performing the mutation analysis, we considered this family to be of intermediate-type CMT, because the median NCV of the proband (III-1; Fig. 2-A) was markedly reduced, whereas that of her mother (II-4; Fig. 2-B) was not reduced. However, it was subsequently revealed that the heterogeneous phenotypes were due to different genetic defects: a duplication of 17p11.2-p12 was detected in the proband (III-1), whereas a deletion of the same region was found in her mother (II-4). Although the occurrence of CMT1A duplication and HNPP deletion in this family might be purely coincidental, the presence of both of them in the same inherited neuropathy family might have resulted as misdiagnosis of intermediate-type CMT.
In addition, patient 2 (II-1) had a de novo CMT1A duplication, and this was attributed to unequal nonsister chromatid exchange during spermatogenesis.4 The prevalence of de novo duplications in CMT1A is much higher for those of paternal origin than for those of maternal origin, which may be due to the recombination fractions for the region being larger in females.5


 XML Download
XML Download