

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) visualize and estimate quantitatively metabolic, biochemical or functional activities in a living brain; hence, called as functional imaging. Since SPECT and PET were first introduced in Korea in 1986 and 1994 respectively, technical advances and availability have grown at a remarkable pace. As of 2005, there are 187 SPECT cameras, 54 PET or PET/CT, 4 microPET, and 15 cyclotrons (which produce radiolabeled tracers) across the country; moreover, SPECT/CT fusion and high-resolution research tomograph (HRRT) will be introduced. In the future, it may be possible to see high-resolution Compton gamma camera and PET/MR fusion made in Korea. There is no doubt that the rapid expansion of this infrastructure will contribute enormously to efforts of neuroscience community searching for prevention or curative treatment of neurodegenerative disorders - the last frontier of medicine.
The goal of this review is to discuss the current issues on Parkinson's disease (PD), and establish the rationale for using this relatively new technology in clinical research and practice for PD.
DEFINITION OF PARKINSON'S DISEASE
In 1817 at the start of the Industrial Revolution, James Parkinson first reported PD in his monograph, "An Essay on the Shaking Palsy" based on his observations as a physician in East London. In 1888, Jean-Martin Charcot, then prominent French neurologist, pointed out that patients with this disorder do not have real weakness nor always have tremor, and thus, shaking palsy or paralysis agitans is a misnomer. He renamed this disorder as Parkinson's disease. In 1912, Friedrich Lewy, German neurologist in Alois Alzheimer's laboratory, first described Lewy bodies. In 1960, Hornykiewics showed that the dopamine content of the substantia nigra and corpus striatum in postmortem PD brains was extremely low, less than 10% of normal, ushering into an L-dopa era. In 1997, Polymeropoulos et al reported the first genetic cause of PD, mutations in the SNCA (α-synuclein) gene.1 Two months later, Spillantini et al showed that α-synuclein was a major component of Lewy bodies in sporadic PD, linking genetic and sporadic PD.2 It has opened the third era in PD.
The diagnosis of PD has been based on clinical and pathological features. UK PD Society Brain Bank clinical diagnostic criteria (one of most stringent criteria, widely used for research purposes; also known as QSBB criteria) consist of three steps: (1) diagnosis of parkinsonian syndrome; (2) exclusion of atypical features including significant family history and early severe dementia; (3) supportive prospective positive criteria observed during the longitudinal evaluation.3 The clinical diagnosis of PD is confirmed by postmortem pathological examinations with loss of pigmented neurons and the presence of Lewy bodies in the substantia nigra. Based on these criteria, 76% of clinically diagnosed PD was confirmed pathologically.4 This traditional way of defining PD is challenged by mounting new information derived from studies in familial PD.
CHANGING CONCEPT: PD AS A CLINICAL SYNDROME
Since the first report of a familial PD with mutations in α-synuclein gene in 1997, at least five more genes have been identified as a causative mutation in familial PD (Table 1).5 Subsequent clinical and pathological studies of affected people in these families revealed unexpected findings. First, patients with leucine-rich repeat kinase 2 (LRRK2) mutation showed pleomorphic pathology even within a family: Lewy bodies, tauopathy or neuronal loss without intracellular inclusions.6,7 Patients with parkin mutations showed dopamine neuronal loss without intracellular inclusions and/or tauopathy in most cases, and Lewy bodies in some with heterozygous mutation.8 Interestingly, previous pathological studies in clinically diagnosed sporadic PD also reported a few cases showing neuronal loss without intracellular inclusions9 or tauopathy.10 Second, clinical phenotypes and pathological features ranged from typical idiopathic PD to diffuse Lewy body disease between individuals with mutation in the same gene, encoding LRRK27 or α-synuclein multiplication. 11 Third, seemingly sporadic PD might in fact have a genetic basis, blurring the boundary between sporadic and familial PD.12-14 Therefore, significant family history should not exclude the diagnosis of PD,15 as in the current QSBB criteria. These observations also indicate that the presence of Lewy bodies in the postmortem brain cannot be used to "confirm" the disease16 because the pathology of PD is likely an outcome of disease process, i.e. phenotype, rather than a determining factor of the disease.
NEW APPROACH TO PD: A NEED FOR PATHOGENIC CLASSIFICATION
Traditional belief that PD is a relatively homogeneous disease entity, which can be separated from other similar disorders by applying stringent diagnostic criteria, has crumbled. Now, we have definite evidence for the longstanding argument that PD is a clinical syndrome.17,18 More than 100 years ago, Charcot changed the name of this disorder, "shaking palsy" that is a syndromic description, to "Parkinson's disease" that implies a homogeneous entity as a disease. After all, this revised term is turned out to be a misnomer again.
What is the impact of these new developments in everyday practice? Perhaps, the most compelling issue is whether the diagnosis of PD will be good enough for the treatment of disorder. Will this syndromic diagnosis guide us to tell patients the prognosis and definite treatment? From the clinician's viewpoint, it is most useful if a disease is defined and classified based on the pathogenesis. By analogy, the diagnosis of pneumonia is not sufficient for the treatment. The physician should understand the underlying pathogenic process, e.g. infection, autoimmune or aspiration. Then, is it possible to categorize idiopathic sporadic PD based on pathogenesis?
The study of genetics has been successful in identifying key proteins and causative mutations in familial PD. It will continue to make headway in elucidating molecular pathogenesis of familial PD. However, monogenic PD explains only 20% of the early onset PD and less than 3% of the late onset PD.19 The rest are all sporadic and idiopathic PD. Can we then relate findings in familial PD to sporadic PD?
Clinical phenotypes of familial PD, if all subtypes are lumped together, are as heterogeneous as those of sporadic PD. However, it is relatively homogeneous in each subtype of familial PD. The clinical phenotypes of dominant parkinsonism, especially Park8, are largely indistinguishable from typical late-onset idiopathic PD. The phenotype of Park1 is an aggressive dopa-responsive parkinsonism with earlier onset (mean age of 46.5 years) and more rapid progression, compared with typical idiopathic PD.20 It also shows a gene-dose effect on clinical and pathological phenotypes, more extensive with greater multiplication of α-synuclein gene.11 These findings clearly demonstrate that the mutated gene is a major, but not exclusive, factor determining clinical and pathological phenotypes. Mutations in α-synuclein gene are known to cause abnormal lipid and vesicle dynamics,21 while mutations in LRRK2 gene are to cause abnormal MAPKKK (mitogen-activated protein kinase kinase kinase) signaling.22,23
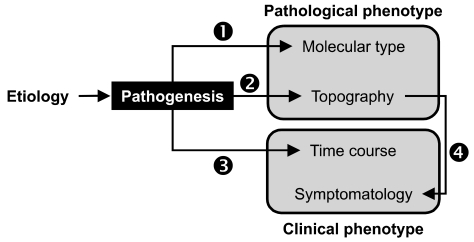
In contrast, the clinical phenotype of recessive parkinsonism (e.g. Park2, Park6 and Park7 with mutations in parkin, PTEN-induced putative kinase 1 (PINK1) and DJ-1 respectively) is typically a young-onset PD (YOPD, the onset between 20~40 years of age) with slow progression and good response to L-dopa. This triad of recessive genes is variably associated with mitochondria, and protective against mitochondrial dysfunction and oxidative stress.24,25 Although the pathological data are not available to all subtypes, at least, Park2 shows neuronal loss generally without protein aggregation, distinct from mutations in dominant genes that produce protein aggregation pathology.25 Therefore, while different gene mutations are associated with different pathogenic mechanisms and different clinical and pathological phenotypes, those with similar pathogenic mechanisms tend to show similar clinical phenotype (Fig. 1).
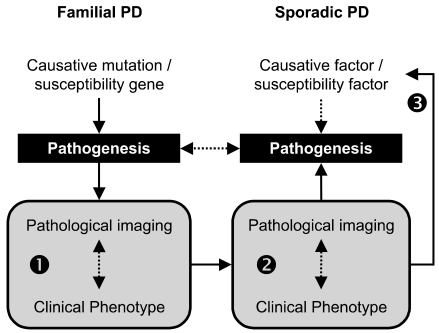
If one accepts the argument that the pathogenic mechanism is a major factor determining clinical and pathological phenotypes, one may assume that the pathogenic mechanism of a subtype of sporadic PD would be similar to that of familial PD with similar clinical and pathological phenotypes. It has been thought that heterogeneity in sporadic idiopathic PD may suggest the existence of subgroups within the disorder, with distinct clinical patterns and perhaps different pathogenic mechanisms.26 However, without a specific biologic marker or a test, the subtyping of sporadic PD has been entirely dependant on clinical features: specific motor symptoms (tremor predominant vs. postural instability and gait difficulty, PIGD), age at onset (young-onset vs. old-onset), accompanying dementia, or data-driven approach without a priori assumption.27,28 Fifteen years ago, Graham et al reported interesting findings in their data-driven approach, which are relevant to the current observations in familial PD. They could identity 3 distinctive subtypes in sporadic PD: the "motor only", "motor and cognitive", and "rapid progression".27 The "motor only" subtype could correspond to benign YOPD in most recessive parkinsonism, whereas the "motor and cognitive" and the "rapid progression" could correspond to Park8 and Park1 (with duplication or triplication) respectively. This example demonstrates that a parallelism may indeed exist between sporadic and familial PD with respect to the disease phenotype. Recently, new application of PET and SPECT for exploring biochemical and pathological aspects of disease,29-31 which will be discussed in the following sections, makes it possible to test this hypothesis by providing necessary biomarkers for sporadic PD (Fig. 2).
CURRENT STATE OF FUNCTIONAL IMAGING FOR FAMILIAL PD
In recent years, PET and SPECT have been increasingly used for studies on familial PD. In 2001, Hilker et al in Germany reported a [18F]-dopa PET study in 5 symptomatic and 5 asymptomatic family members with parkin mutation.32 No significant differences were found between familial and sporadic PD. Interestingly, asymptomatic carriers with heterozygous mutations showed mild but significant reduction of Ki (by about 18%) without a rostrocaudal gradient. In the following year, UK Hammersmith group reported two serial observations over up to 10 years: 4 symptomatic parkin patients showed slower progression than sporadic PD, whereas 4 asymptomatic parkin carriers (2 had serial scans) showed mild reduction of Ki but without significant progression.33 In 2003, a French study reported no significant differences in [18F]-dopa findings between 19 parkin patients and 6 parkin-negative, sporadic YOPD.34 In 2005, the UK Hammersmith group confirmed their earlier observations in asymptomatic parkin heterozygotes, showing mild reduction of Ki without anteroposterior gradient in the striatum.35
[18F]-dopa PET study for patients with PINK1 mutations also showed significant reduction of [18F]-dopa Ki in the striatum, more so in the putamen.36 As in parkin mutations, asymptomatic carriers with heterozygous PINK1 mutation showed mild reduction of Ki without significant anteroposterier gradient. Recent findings of [123I]-β-CIT SPECT in a patient with PINK1 homozygous mutation was similar to those of [18F]-dopa PET.37 Recent [18F]-FP-CIT PET study in a Turkish family with DJ-1 mutations showed significant reduction of [18F]-FP-CIT binding in the striatum with significant rostrocaudal gradient in a patient with homozygous mutation, but mild reduction only, without significant rostrocaudal gradient in asymptomatic sister with homozygous mutation.38
Mutations in the LRRK2 gene cause autosomal dominant parkinsonism that is clinically and pathologically indistinguishable from typical late-onset, sporadic idiopathic PD.7,39 Moreover, the disease penetrance in LRRK2 carriers is age-dependent, increasing from 17% at age 50 years to 85% at age 70 years,40 simulating the age-dependent increase of sporadic idiopathic PD. [18F]-dopa PET in two patients with LRRK2 mutation did not show significant differences from idiopathic PD.41 The collaborative study between people in Vancouver and Mayo Jacksonville, using three presynaptic tracers of [18F]-dopa, [11C]-methylphenidate (MP, for DA transporter) and [11C]-dihydrotetrabenazine (DTBZ, for VMAT2 in the synaptic vesicle of monoaminergic terminals), showed similar findings between 4 symptomatic patients with LRRK2 mutation and sporadic idiopathic PD. Two asymptomatic carriers showed mild reduction of [11C]-DTBZ and [11C]-MP binding, but not [18F]-dopa Ki, suggestive of possible compensatory changes in the preclinical stage.42 [123I]-ioflupane SPECT study in 10 Italian patients with LRRK2 mutation (7 familial, 3 sporadic) also did not show significant differences between PD with LRRK2 mutation and without mutation.43
FUTURE PROSPECTS: PATHOLOGICAL IMAGING
Functional imaging in familial PD has been conducted using only dopamine tracers. Heterogeneity of familial and sporadic PD is, however, largely accounted for by non-dopamine and Alzheimer pathology; thus, PET or SPECT with multiple tracers can explore the extent of chemical or morphological pathology in the brain. There are three potentially useful tracers, which are in line with our interests.
The first tracer is a cholinergic ligand. Dementia is the most important variable in the categorization of PD, and attributed to multiple mechanisms including cortical cholinergic deficits,44-46 Alzheimer pathology and cortical Lewy pathology in addition to dopamine or other monoamine deficits. 11C-labeled acetylcholine analog is a presynaptic marker, measuring the activity of acetylcholine esterase in the cortex (which reflects cholinergic terminal density). There are two types: [11C]-MP4A (methylpiperidin-4-yl acetate) developed in Japan, and [11C]-PMP (methyl-piperidin-4-yl propionate) developed in Ann Arbor, USA. Both have similar biological characteristics. PET studies using one of these ligands have been reported for patients with Alzheimer disease (AD),47-49 PD and PSP,50 PD with dementia,51 and subjects with MCI.52 These ligands are affected by acetylcholine esterase inhibitors, such as donepezil, which can be a problem for patients on this drug.53 However, [123I]-IBVM (iodobenzovesamicol), SPECT tracer, binds to acetylcholine vesicular transporter;54 thus, is not likely affected by acetylcholine esterase inhibitors. A SPECT study using this tracer in AD and PD patients was reported in 1996.54
The next tracer of interest is [11C]-OH-BTA-1, commonly called as Pittsburgh Compound-B (PIB). This PET tracer is a derivative of thioflavin-T, which is amyloid-binding histological dye.55 In 2004, Klunk et al in Pittsburgh reported marked [11C]-PIB binding in the association cortex of AD patients, which inversely correlated with [18F]-FDG (fluorodeoxyglucose) uptake.29 Postmortem pathological studies have reported that Alzheimer pathology is common in most, but not all,56 PD patients with dementia.57,58 The natural history of Alzheimer pathology in relation to Lewy pathology is not known.59 At present, [11C]-PIB PET studies for PD with dementia and dementia with Lewy bodies are being conducted in some centers. SPECT tracer for amyloid plaque is currently under development.
The third tracer of interest is [11C]-PK11195, a PET ligand originally developed for the peripheral benzodiazepine binding site (PBBS).60 Since PBBS expresses in activated microglia, it began to be used for neurodegenerative disorders after McGeer reported abundant activated microglia in the PD and AD brains.61,62 This tracer is potentially important because microglial response may be used as a marker for disease activity in PD, which is otherwise not available. Therefore, in principle, it may localize the area of brain where the disease process is active, and also may show the time course of disease activity in a given region - findings which would be highly meaningful for the study of putative neuroprotective agents. In 2005, Ouchi et al first reported a study in PD patients, using two tracers of [11C]-(R)-PK11195 and [11C]-CFT (carbomethoxy-3 beta-(4-fluorophenyl) tropane; DAT ligand).30 They found increased binding of [11C]-(R)-PK11195 in the PD patients, particularly in the midbrain, which correlated inversely with [11C]-CFT binding in the putamen. In the following year, the UK Hammersmith group reported a longitudinal [11C]-(R)-PK11195 PET study in PD patients, which did not show significant changes over 2 years.63
In summary, with a variety of tracers that can provide biochemical and molecular aspects of pathology, we are now in a position to explore longitudinal changes of pathology in PD patients. The "functional" imaging is evolving into the "pathological" imaging; thus, for the first time since James Parkinson's clinical report, it becomes a reality to combine the two major expressions of disease, i.e. clinical and pathological phenotypes, in real time.
CONCLUSIONS
Throughout the history, advances in scientific technology have led to new discoveries and innovative treatments in medicine. With a rapid development of computed imaging device and radiopharmaceuticals, we are now facing a new opportunity for PD, almost 200 years after first reported. Functional imaging will make it possible to categorize PD into subtypes with distinct pathogenesis. It will be also instrumental for clinicogenetic studies identifying susceptibility factors and at-risk groups, which are essential for successful neuroprotective treatment when it becomes available.

 XML Download
XML Download