

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Huntington's disease (HD) is an adult-onset, chronic and progressive neurodegenerative disease. It is clinically characterized by abnormal choreic involuntary movements and by psychiatric, psychological and intellectual disorders, and radiologically characterized by striatal atrophy of variable degree. Pathologically, in atrophied striatum, the normally predominant small projecting neurons are specifically affected. Since these neurons are inhibitory in function, whose long axons terminate in the substantia nigra and use γ-aminobutyric acid (GABA) as a neurotransmitter, GABA levels in the substantia nigra of HD are markedly low.1 On the other hand, dopaminergic nigral neurons remain intact in HD and, indeed, the dopamine level in the HD striatum is higher than normal.2 Therefore, HD is regarded as a relatively dopamine-predominant disease. In agreement with this finding, anti-dopaminergic drugs are clinically effective against choreic movements.
Genetically, HD is an autosomal-dominantly inherited disease. Genetic linkage analysis using DNA fragment as gene marker revealed in 1983 that HD gene may be located at the tip of the short arm of chromosome 4.3 Ten years later, the causal HD gene was finally identified as IT15, which has CAG repeats in the open reading frame (ORF, exactly, in exon 1) just following the 5'end of the gene.4 Since CAG is a codon corresponding to glutamine, the gene product, huntingtin, should have glutamine stretch or chain. Although the function of the normal huntingtin is not fully elucidated, the abnormally long glutamine chain in the mutant huntingtin distorts the third structure of huntingtin protein. Furthermore, these structurally abnormal proteins tend to aggregate and to form inclusion bodies inside neurons or nuclei.5 In addition, mutant huntingtin may be toxic to functionally important proteins in neurons.6
The progress in medical research particularly from the molecular aspect of HD is extremely rapid and promising. Although the clinical application is still far from reality, cell therapy or gene therapy of HD are no longer in the dream but actually a reality in laboratories around the world at present. It is relevant, therefore, to summarize the progresses particularly in therapeutic approaches to HD.
Symptomatic/palliative therapies for HD
1. Drugs that reduce choreic movements and psychiatric disorders mostly based on classical theories
Since Thomas Perry and his group in Canada reported that HD was a deficiency of GABA in the basal ganglia,1 clinically available drugs related to GABA were used in patients to establish their possible anti-choreic effects. However, none was found to be definitely effective against choreic movements.7-11
Based on a recent theory on the mechanism of choreic movements involving hyperactivity of the cortico-striatal glutamatergic pathways, one of the NMDA-receptor antagonists, amantadine was clinically evaluated, but no positive effects were confirmed following oral administration.12
In contrast to the above mentioned rather disappointing results mostly obtained in theoretically supported clinical trials, antipsychotic or dopamine-depleting drugs were confirmed largely in 1970's13 to be effective not only for choreic movements but also for psychiatric disorders associated with HD. Later, this was found to coincide well with the relatively dominant dopamine compared with other neurotransmitters in HD striatum.2 Attention has recently focused on the antichoreic effects of tetrabenazine, which selectively depletes monoamines by reversibly binding to the type 2 vesicular monoamine transporter.14
A double-blind, randomized, placebo-controlled clinical study of ethyl-eicosa-pentaenoic acid (ethyl-EPA) on choreic movements of HD revealed that this agent exerts subtle beneficial effects on motor dysfunction of HD.15 Since adverse events of ethyl-EPA were negligible, the potential use of ethyl-EPA in HD is warranted.
2. Stereotaxic surgery for choreic movements
Stereotaxic surgery is rarely used in HD. The target is the internal segment of the globus pallidus,16 which is the origin of the final common path; ansa lenticularis. Usually the outcome is not disappointing, though it is rather transient because the disease is highly progressive.
3. Repetitive transcranial magnetic stimulation (rTMS)
A beneficial effect of rTMS on HD-related choreic movements was first proposed by an Italian group in 1994.17 However, although small scale investigation was carried out,18 only little information is available about the effects of rTMS in HD. Moreover, the mechanism of action of rTMS, if any, is not clear at present.
Anti-degenerative therapies for HD
1. Anti-excitotoxicity therapy
Based on the excitotonic theory of striatal neuronal degeneration in HD, clinically available anti-excitotoxic drugs such as amantadine, memantine and riluzole, were used in HD patients to suppress choreic movements. Although riluzole (which reduces striatal glutamate release) was reported to ameliorate the intensity of choreic movements,19 there is no strong clinical evidence to support the hypothesis.
2. Reversal of mitochondrial dysfunction
Although the debate on the relationship between presence of mutant huntingtin and mitochondrial dysfunction is still unsettled,20 coenzyme Q10 (CoQ10), an essential cofactor of the electron transport system, was clinically tested in HD in several centers. However, a recently completed large-scale double-blind, randomized, placebo-controlled clinical trial of CoQ10 (at 300 mg twice daily) concluded that there was no significant slowing of functional decline in patients with early HD.21
Another therapeutic candidate for energy crisis is "creatine". Creatine is a substrate for the enzyme creatine kinase and can protect vulnerable neurons from degeneration through ATP generation.22 Since a rather large dose of creatine administered to HD patients was well tolerated and resulted in reduction of serum 8-hydroxy-2'deoxyguanosine level, an indicator of oxidative stress,23 long-term clinical trials are warranted indeed.
3. Anti-apoptosis therapy
In a mouse model of HD, minocycline, a tetracycline-derivative compound, delayed HD disease progression through down-regulation of apoptosis-related caspase-1 and -3, and nitric oxide synthetase activities.24 Open clinical trials of minocycline in HD based on the abovementioned finding revealed a long-lasting beneficial effect on HD in terms of both choreic movements and psychiatric disorders.25 Again in the mouse model of HD, the endogenous hydrophilic bile acid, tauroursodeoxycholic acid, resulted in reduction of apoptosis in striatal neurons, and improvement in locomotion and sensorimotor deficits.26 Although basic research in this field is active at present, no results of large clinical studies have been reported.
Recently, attention was focused on the antibiotic rapamycin due to its anti-aggregate formation and neuroprotection (vide infra). In addition, rapamycin was found to have anti-apoptotic effect as well.27 Since rapamycin is already used clinically as an immunosuppressive agent in the United States, this or related analogues are suitable candidates for therapeutic investigation in HD.
Restorative/reparative therapies for HD
1. Neural trophic factors
The brain-derived neurotrophic factor (BDNF) localized in the cortico-striatal pathways may be one of the targets of mutant huntingtin, and is reduced in the HD brain to less than a half of the normal level.28 Indeed, mutant huntingtin suppressed the levels of both mRNA and protein of BDNF in HD mouse model.28 Although the effects of glial cell-line derived neurotrophic factor (GDNF), delivered by lentivirus, were negative,29 fibroblast growth factor-2 (FGF-2)30 and ciliary neurotrophic factor (CNTF) were reported to induce BDNF-like positive results in HD model.31 Further studies are needed including drug delivery systems before next phase of clinical trials. Although neural trophic factors are not specific in a particular disease, they may contribute to neural maintenance and survival, and may be therapeutically useful in neurodegenerative diseases including HD.
2. Fetal tissue or primary fetal cell transplantation
Premordial striatal tissue grafting into the striatum of ibotenic acid-induced rat HD model was first introduced by Isacson et al in 1985,32 suggesting that grafting can result in recovery of the neurotransmitter systems. The basic idea was fostered to clinical application, and a 6-year follow-up study of 5 patients who each received implantation of the ganglionic eminence of the human embryo into the head of the caudate nucleus and putamen, was presented in 2006.33 According to this study, the transplantation did not result in deterioration of chorea, and cognitive performance measured by non-timed tests remained stable in 3 out of 5 patients. Two patients did not show any beneficial effects. However, transplantation using fresh fetal tissues is always limited by not only technical but also ethical problems. Therefore, although the clinical outcome was not disappointing, this line of study will shift to stem cell transplantation as discussed below.
3. Stem cell transplantation
Theoretically, "stem cells" are one of the most hopeful tools for radical cure of neurodegenerative diseases including HD. It is anticipated that neural stem cells from fetal brain or adult brain can differentiate effectively into striatal neurons and expand over a long period of time. Although fetal brain cells have been isolated and maintained in the form of neurospheres, attempts to differentiate them into striatal neurons have not yet been successful.34 While it is good news that neural stem cells are located in the subventricular zone of the adult human brain, their functional role in the living brain and their availability for treatment of the diseased brain is still unclear.
In this respect, Lee et al35 demonstrated that intravenous administration of human neural stem cells in a quinolinic acid-induced HD rat model resulted in accumulation of cells in and around the striatal lesion and their differentiation not only to glial cells but also to neurons as well.35 Moreover, experimental rats exhibited a shift to normal response in apomorphine-induced rotation test, and showed reduced striatal atrophy compared with the control rats that received quinolinic acid injection only. The functional recovery of rat behavior and the normalization of brain pathology are promising and need to be further confirmed in the near future.
So far, no study has claimed the differentiation of embryonic stem cells into striatal neurons, and this seems far from clinical application.
Molecular targets in specific and radical therapies for HD
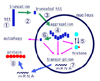
1. Short summary of molecular mechanisms of neural degeneration in HD
The HD gene is localized on chromosome 4p16.3 and named "interesting transcript 15 (IT15)". The human and murine HD genescontain 67 exons. The CAG repeats are located at the region near the 5'-end of the open leading frame, which encodes a large protein of 350 kDa, and is named "huntingtin".4 The number of CAG repeats in normal chromosome is around 17, at least less than 34, whereas that in HD chromosome is more than 36, mostly more than 40. These numbers of CAG repeats are unstable and inversely correlate with the age at onset and with the severity of symptoms. Since CAG is a corresponding codon to glutamine, mutant huntingtin has abnormally long glutamine stretch near the N-terminal end. Huntingtin protein is ubiquitously expressed throughout the central nervous system, including every neuron. In the HD brain, small projecting neurons located in the caudate and the putamen are most vulnerable. However, the mechanism of this selectivity is not yet elucidated (vide infra).
As mentioned in the Introduction, HD is an inherited disease with autosomal dominant trait. Since the phenotypes of rare homozygote patients are almost the same as those of heterozygotes, HD may be caused by a gain-of-function mechanism. Indeed, the truncated mutant huntingtin easily enters the nucleus,36 aggregate to form the β-sheet,5,37 and recruits many types of proteins such as histones, splicosomes,38 enzymes and transcription factors,39 which are essential for cell survival. Therefore, it is possible that the mutant huntingtin is at least in part neurotoxic. It is also possible that mono- and oligomer of mutant huntingtin are much more harmful than aggregates themselves.6 Furthermore, recent studies have shown that BDNF located in the cortico-striatal pathways may be lost when normal huntingtin is suppressed in the adult animal, and the loss of BDNF function may contribute in part to the striatal neuronal degeneration.40 Therefore, a loss-of-function mechanism may also occur as well in the HD brain.
Fig. 1 is a summary of therapeutic strategies based on the understanding of molecular mechanisms of neurodegeneration happened in HD brain.
2. Inhibition of truncation of huntingtin
As mentioned above, the very first step of neurotoxicity of mutant huntingtin is truncation of huntingtin, which occurs in the cytoplasm by caspase-2,-3,-6, calpain and/or aspartic endopeptidase. Since there is no specific inhibitor to date against the latter two enzymes, only caspase inhibitors (z-VAD-fmk and z-DEVD-fmk) have been tested. These inhibitors blocked cleavage of huntingtin and subsequently reduced cytotoxicity.41 Recently caspase-3- and -6-resistant huntingtin gene-containing YAC expressing mice were generated. Interestingly, behavior and brain pathology in these mice were reported to be less severe in caspase-6-resistant mice than wild type.42 However, caspase does not only cleave huntingtin but also many other proteins including those protecting cells from degeneration. Therefore, it may be extremely difficult to find a therapeutic window even at the pre-clinical phase.
3. Inhibition of aggregate formation
Soon after the causal gene was identified and just before the nuclear inclusion body was identified, transglutaminase drew attention as a candidate pathogenic factor that interfere with the normal functions of cellular proteins, due to its molecular cross-linking activity.43 After wide acceptance of the importance of the aggregates, much more attention was paid to this enzyme. Particularly, Chun et al44 demonstrated that transglutaminase specifically interacts with a truncated form of mutant huntingtin. Indeed, intraperitoneal injection of cystamine, a transglutaminase inhibitor that is converted to the potent antioxidant cysteamine in vivo, in transgenic mouse model of HD resulted in improvement of survival rate and reduction of tremor-like abnormal movements.45 Moreover, PET imaging and histological studies of HD mouse model after cystamine treatment demonstrated the beneficial effects of cystamine not only in reduction of aggregates but also in protection against striatal neuronal death.46
As discussed briefly above, one question is not yet settled are intraneuronal aggregates/inclusion bodies harmful to cell survival; conversely, are they protective for cell death by sequestering harmful free forms of mutant huntingtin molecule? Interestingly, the same research group presented data in support of the former concept in one publication, and supported the latter concept in the another.47,48 Nevertheless, many studies identified small moleculesas potential polyglutamine aggregation inhibitors through screening systems.47, 49-51 Indeed. Nukina et al51 from RIKEN reported that trehalose, a popular disaccharide, was one candidate inhibitor of polyglutamine-mediated protein aggregation, and actually reduced symptoms of HD mouse model. Similarly, Congo red binds to the β-sheet structure and inhibits polyglutamine oligomerization.52 Several other compounds such as CHIP,53 Y-27632,50 PGL-135,54 Juglone and Celastrol55 are under investigation both in vitro and in vivo, though no solid results have yet been published.
The truncated huntingtin was demonstrated to interact with heat shock proteins, Hsp40 and Hsp70, in huntingtin-expressing cultured cells in a polyglutamine chain length-dependent manner. This means that these molecular chaperones may be effective in suppressing huntingtin aggregation.56 Since the anti-cancer drug, geldanamycin, is known to bind specifically to chaperone Hsp90 and to inhibit its chaperone function,57 Wanker's group in Germany applied geldanamycin at nanomolar concentrations to huntingtin-expressing cultured cells and found that geldanamycin induced Hsp40, Hsp70 and Hsp90 and suppressed aggregate formation.58 In addition, chaperone manipulation by geldanamycin in mice model delayed aggregate formation for one week.59 Thus, pharmacological induction of chaperones seems to be subtle, but worth further investigation.
4. Normalization of transcriptional dysregulation
Just after the causal gene of HD was identified in 1993, Gerber et al pointed out that based on the existence of the CAG repeat in huntingtin gene, huntingtin could interact with transcription factors.60 Indeed, recent studies have revealed that mutant huntingtin directly interacts with various nuclear transcription factors61 and interferes with the normal transcriptional process in vulnerable neurons,62 leading to neuronal degeneration. The discovery of specific component(s) of the transcription apparatus caused by mutant huntingtin using newly developed assay system63 should allow the use of such molecules for normalization of the transcriptional dysregulation and thus be applied clinically in the future.
Another approach to the transcriptional dysfunction emerged from a study using a Drosophila model of HD. Although the transcription processes in cells are balanced between histone acetylation and deacetylation, this reaction may be shifted to deacetylation catalyzed by histone deacetylase (HDAC) in HD model and probably in polyglutamine diseases.64 Correction of this shift could be, therefore, a suitable strategy for a novel therapy of HD. Indeed, one of the HDAC inhibitors, sodium butyrate, was reported to modulate the transcription, significantly enhanced survival, and delayed the neuropathological changes in a R6/2 HD mouse model.65
5. Enhancement of autophagic clearance of abnormal proteins
The lipophilic macrolide antibiotic rapamycin was recently reported to induce autophagy by inactivating "mammalian target of rapamycin (mTOR)" which is a type of serine-threonine kinase.66 Rubinsztein's group in Cambridge, UK, demonstrated that high glucose concentrations in cultured cells enhanced clearance of mutant huntingtin, together with increased autophagy and reduced phosphorylation of mTOR.67 This line of evidence was also presented in fly and mouse models of HD.68 In contrast to this "rapamycin-related story", Yamamoto et al69 recently claimed the presence of mTOR-independent autophagy in huntingtin-expressing cultured cells, and that such autophagy could be achieved by insulin-signaling pathway.69 Although these findings are still in the laboratory phase, an enhanced clearance of mutant huntingtin is a reasonable direction in developing a new therapy for HD.
6. Specific inhibition of huntingtin gene expression by RNA interference
Although a loss-of-function mechanism may exist, we believe that the major mechanism of the striatal neural loss is due to a gain-of-function. This concept is supported by many studies.6,36,39,40 If so, as Haque et al70 has suggested first, down-regulation of mutant huntingtin expression may be a promising therapeutic strategy. This concept was strongly supported by a recent publication by Yamamoto and colleagues71 using a conditional mouse model of HD. In their study, they demonstrated that "switching off" of the transgenic huntingtin expression at the adult age lessened the severity of the disease, suggesting that HD might be reversible.
Moreover, we have found in our preliminary single neuron analysis that the ratio of mutant to normal huntingtin gene expression of the most vulnerable striatal remaining neurons in a HD patient was 10-15% more than that of Purkinje cells, which are the least affected in HD (Fig. 2). It is reasonable to suppose that neurons would be killed earlier if a relatively large amount of mutant huntingtin is expressed even if the difference is subtle. This assumption can also explain the selectivity of striatal neuronal death in HD. Therefore, in order to formulate a molecular therapeutic strategy for HD, it is feasible to suppress the mutant huntingtin expression. In this respect, several techniques have already been explored to suppress the expression of huntingtin, including antisense technology72,73 and catalytic DNA.74 However, these tools have their own problems such as instability of the single strand RNA molecule or unexpectedly low efficacy.
In this respect, it is worthy to note that RNA interference (RNAi), the process of posttranscriptional gene silencing mediated by double-stranded RNAs (dsRNAs) first discovered in Caenorhabditis elegans,75 has rapidly emerged as an important tool for regulating the gene expression in a wide range of species76 including mammals. Particularly inmammalian cells, inhibition of the specific gene expression is mediated by short inhibitory RNAs (siRNAs), which are dsRNAs composed of 21-22 nucleotides (nt) with 2 nt overhang on 3' terminus of each strand, whereas dsRNAs longer than 30 nt induced a global suppression of the gene expression by a mechanism related to interferon response.77,78 Therefore, several studies, including those from our laboratories, have started to develop RNAi systems designed to suppress huntingtin expression.
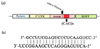
In order to prepare 21 nt RNAs, we first searched BLAST through the HD mRNA and found two sites that have unique sequences as valid targets of siRNA siRNA-5'UTR in 5' untranslated region and siRNA-HDExon1 in ORF at immediate upstream of CAG repeats. Comparison of the silencing effects of these two siRNAs using cultured cells showed that siRNA-HDExon1 is much stronger than the other.79 Indeed, the lowest concentration of the effective siRNA-HDExon1 to suppress huntingtin gene expression in COS-7 cells was 5 nM.79 The nucleotide sequence of the siRNA-HDExon1 is shown in Fig. 3, and its effect at 40 nM is shown in Fig. 4. Since other genes such as β-actin and GAPDH were not altered, siRNA-HDExon1 specifically suppresses the expression of huntingtin gene.79 At this stage, these studies are ready to move to the in vivo experiment.
SiRNA-HDExon1 was introduced intraventricularly into a R6/2 mouse model of HD on the second day after birth. The injected siRNA-HDExon1 inhibited transgenic huntingtin expression in brain neurons and reduced the number and size of intranuclear inclusions in striatal neurons (Fig. 5). More importantly, siRNA treatment significantly prolonged longevity (Fig. 6), improved motor function, and slowed down the loss of body weight.80
Similar in vivo effects of double-stranded RNA designed toward silencing the huntingtin gene were reported by other groups.71-83 However, these reports are different from our results at least with regard to the following three points; 1. nucleotide sequences of dsRNA used were different. 2. short hairpin RNA (shRNA) was used, instead of straight siRNA, in order to stably and constantly produce siRNA in cells. 3. those shRNAs were ligated to vectors of adeno-associated virus (AAV), instead of naked siRNA.
In May 2006, an important study was published in Nature, which reported that administration of shRNAs with AAV vectors to produce long-term suppression of the target gene could interfere with the normal function of endogenous microRNAs. This may be due to oversaturation of endogenous microRNA/exogenous shRNA pathways in which both RNA molecules use the same components of RNAi apparatus.84 Potential problems that could also occur in RNAi were also recently pointed out by Aronin.85 We strongly intended, from the very first step of our study, to avoid unexpected and uncontrollable adverse events in volunteer patients. Therefore, it is important to develop a "transient" suppression system of huntingtin gene as we did, instead of long-term suppression. Since the rate of expression of AAV-delivered shRNA in cells can not be controlled, I absolutely prefer to use siRNA that will remain only transiently inside the cells, even if siRNA should be delivered directly into the striatum or ventricles of HD patients.
Finally, the result of an interesting study should be mentioned here, which was based on the understanding of HD as a disease of metal abnormality. Indeed, Nguyen et al86 have applied the metal-binding compound clioquinol to R6/2 mouse model of HD and revealed almost the same level of clinicopathological beneficial effects. It is, therefore, important to screen available chemicals as many as possible, in order to find beneficial effects in animal models of HD, and hopefully in human.
CONCLUSION
In 1972, almost 100 years after the first description of HD by George Huntington, a GABA deficiency in HD brain was discovered. In 1983, nearly 10 years after that, a localization of HD gene was found at the short arm of the chromosome 4. Furthermore, in 1993, exactly 10 years after that, the causal gene of HD was finally identified. From analogy to this previous history of HD research, we hoped that in 2003, 10 years after the identification of the HD gene, a radical treatment could be clinically realized. Unfortunately, this is not the case. However, I am pleased to know that from around 2003 we can discuss about "therapeutic strategies" for HD with reality. Although we have a long way to go in developing an actual radical treatment for HD, I hope that the novel strategies including a stem cell therapy and/or a gene silencing therapy could reach to the level of clinical trials in near future.




 XML Download
XML Download