

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Acquired ocular motor apraxia is characterized by loss of voluntary control of saccades and smooth pursuit, although reflexive eye movements are preserved.1,2,3
Patients with the most severe form of acquired ocular motor apraxia exhibit fixation of gaze. Saccadic and smooth pursuits are rare or often absent, but the vestibulo-ocular reflex and quick phases of vestibular nystagmus are preserved. This rare syndrome must be distinguished from various saccadic abnormalities present in several degenerative diseases involving the basal ganglia. Acquired forms of ocular motor apraxia have been reported in bilateral lesions affecting both the posterior cerebral hemisphere and the posterior part of the frontal lobe including the frontal eye field (FEF).3,4 However, ocular motor apraxia after bilateral subcortical infarctions has not been reported. We report a patient with acquired ocular motor apraxia due to bilateral basal ganglia infarcts without cortical lesions.
CASE REPORT
A 60-year-old man was admitted to our hospital because of sudden speech disturbance and dysphagia that had begun suddenly a few hours previously. He had developed dysarthria and right-side weakness 6 years previously, which had improved to mild dysarthria at the time of admission. Other medical history included untreated hypertension and 80 pack year smoking. Neurological examination showed alert mental status with cooperative behavior, and comprehension was intact. Cranial nerve examination showed facial diplegia with severe dysarthria and dysphagia, and an inability to protrude the tongue. Light reflex was intact and pupils were equal, and visual acuity and confrontation visual fields were normal. When the patient was asked to fixate on an object in the peripheral visual field, he could not follow the object with his eyes and instead rotated his head toward the object. Extraocular movements showed no voluntary gaze in the horizontal and vertical planes. Intermittent involuntary rightward saccades and smooth pursuit were observed, but no ocular movements were induced during voluntary gaze. The vestibulo-ocular reflex was intact. He also had difficulty in opening both eyes upon command. He could direct his hand to an object with either arm and could recognize complex visual scenes presented on the illustrations. Other motor, sensory, and cerebellar functions were intact.
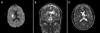
Over the following several days, the difficulty he had in opening his eyes improved; similarly, the rightward voluntary gaze also slightly improved, with intermittent voluntary saccades possible. However, the vertical and leftward horizontal saccades and smooth pursuit remained unchanged. Magnetic resonance imaging of the brain showed acute infarction in the right caudoputaminal area and chronic infarction in the left putamen with Wallerian degeneration in the left pyramidal tract (Fig. 1). No other lesion was observed in all sequences including diffusion-weighted imaging. He was prescribed aspirin and clopidogrel, and discharged with slight improvement. At a 3 month follow-up examination after stroke onset, he still showed severe dysarthria and dysphagia. However, the extraocular movements had improved markedly, rightward saccades and smooth pursuit were almost normal, but catch-up and slow-velocity saccades were observed during attempted leftward gaze. However, voluntary vertical saccades and smooth pursuit remained unchanged.
DISCUSSION
Acquired form of ocular motor apraxia has rarely been reported in bilateral frontal or frontoparietal lesions, and also reported as a complication of cardiopulmonary bypass.5
Defective voluntary eye movements probably reflect disruption of the descending pathways from both the FEFs and the parietal cortex. After sequential bilateral subcortical infarction, our patient showed marked ocular motor apraxia and pseudobulbar palsy with transient apraxia of eye opening. Acquired ocular motor apraxia has rarely been reported in bihemispheric lesions,3,4 but marked ocular motor apraxia in bilateral subcortical lesions has not been reported.
The proposed mechanism of ocular motor apraxia in our patient is bilateral disturbance of the oculomotor circuit at the level of the basal ganglia. Three cortical areas may trigger saccades: the FEF, the supplementary eye field, and the parietal eye field. The oculomotor circuit originates in the FEF and posterior parietal cortex (PPC). The FEF, located in the posterior portion of the middle frontal gyrus, controls intentional saccades. The PPC, located in the intraparietal sulcus in humans,6 is mainly involved in triggering reflexive saccades7 and smooth pursuit.8 The fibers then project to the body of the caudate nucleus, dorsomedial globus pallidus and ventrolateral substantia nigra,9 and terminate in the premotor reticular formations of the brainstem. We postulate that bilateral caudoputaminal infarctions in this patient disrupted the bilateral oculomotor circuit from FEF and PPC and resulted in saccadic and pursuit disturbance.
Until now, it has been widely accepted that only bilateral cortical lesions affecting the cortical eye areas result in visible saccade disturbances.10 Similarly, it is widely accepted that subcortical lesions involving the putamen and pallidum result in subtle impairment of intentional saccades,11 which can only be ascertained using eye movement recordings. Eye movements are disturbed in most degenerative cerebral diseases involving the basal ganglia. Saccadic intrusions and impairment of antisaccade task are frequently observed in Huntington's disease, and vertical saccades are typically slowed in the early stages of progressive supranuclear palsy.12 But, total impairment of saccades and smooth pursuit are only observed in the terminal stages of the disease.
Despite the above, our patient showed marked disturbance of voluntary saccades and smooth pursuit, features typical of acquired ocular motor apraxia. He showed asymmetric ocular motor apraxia that was more severe in leftward gaze than in rightward gaze. This feature suggests that, to develop ocular motor apraxia, the bilateral oculomotor circuit must be damaged, and that the unilateral oculomotor circuit dominates the contraversive control of saccades and smooth pursuit.
Another characteristic feature of our case was concurrent appearance of severe pseudobulbar palsy and transient lid opening apraxia, the latter being a disabling syndrome characterized by an inability to open the eyes at will. The combined pseudobulbar palsy indicates disruption of the bilateral corticobulbar tract and suggests proximity of the oculomotor circuit to the corticobulbar tract. Lid opening apraxia is associated with extrapyramidal diseases, acute strokes involving the nondominant hemisphere, acanthocytosis, Wilson's disease, and blepharospasm.13 The pathomechanism of lid opening apraxia and its relationship with ocular motor apraxia remain unknown. Only one case of concomitant lid opening apraxia and ocular motor apraxia has been reported to date.14
We postulate bilateral disruption of the corticosubcortical oculomotor pathway as the cause of ocular motor apraxia in our patient.
 XML Download
XML Download