

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
LMNA-related muscular dystrophy is a rare disease with variable myopathic phenotypes and for which a causative mutation in the lamin A/C gene (LMNA) has been identified.1 Emery-Dreifuss muscular dystrophy (EDMD) has been known as a phenotype of LMNA-associated muscular dystrophy disorders, along with limb-girdle muscular dystrophy type 1B (LGMD1B) and LMNA-related congenital muscular dystrophy (L-CMD). EDMD is characterized by a clinical triad of early contracture, humeroperoneal distribution of muscle weakness, and cardiac conduction block.2 EDMD symptoms typically present in childhood, and can appear before the age of 2 years.3 Diagnosing early-onset EDMD is challenging because the characteristic muscle weakness pattern and contracture do not appear definitively when the first symptoms appear. Moreover, histopathologic findings in LMNA-related muscular dystrophy are known to show nonspecific myopathic changes such as size variations in muscle fibers, increased internal nuclei, and type 1 fiber predominance without dominant dystrophic patterns.4
While the clinical manifestations of LGMD1B often overlap with those of EDMD, some features can be used to differentiate the two phenotypes. Contracture involvement is not an early sign and is often mild in LGMD1B.5 The pattern of muscle involvement is also milder in LGMD1B, with a proximal limb-girdle distribution and distal leg weakness. Delayed diagnosis is more common in LGMD1B patients due to the later involvement of contracture.6 Nonspecific symptoms in childhood also often complicate the ability to differentiate LGMD1B from other types of limb-girdle muscular dystrophy.
Due to cardiac and pulmonary involvement in the disease course of laminopathy, early diagnosis and periodic cardiac evaluation are important for preventing life-threatening complications. These risks have led to recent studies recommending early genetic tests including exome sequencing in patients with undiagnosed and suspected muscular dystrophy with contracture.78 In addition, consecutive neurologic examinations should be emphasized to monitor the involvement of additional symptoms. However, due to the benign progression of muscle weakness, undiagnosed laminopathy patients are likely to be lost in follow-up, thus reducing the probability of periodic cardiac evaluation.
In this study, we reviewed four patients who were diagnosed with other types of muscular dystrophy or unspecified myopathy at first symptom presentation between the ages of 2 and 5 years, but who were recently rediagnosed with LMNA-associated muscular dystrophy by genetic screening. We evaluated the factors that hindered the early clinical diagnosis in these patients.
METHODS
Patient selection and LMNA mutation detection
We recently performed targeted sequencing of 69 selected genes associated with myopathy in the idiopathic muscular dystrophy patient cohort in our hospital.9 Patients were selected by applying the following inclusion criteria: 1) muscular dystrophy, congenital myopathy, congenital muscular dystrophy, metabolic myopathy, or distal myopathy based primarily on clinical phenotypes and secondarily on pathologic findings (if available), 2) genetically undetermined myopathy, and 3) availability of blood or muscle samples. Genomic DNA was extracted from either blood or muscle. A customized panel containing all coding exons and exon–intron junctions of 69 genes that are known to be responsible for myopathy was used, and LMNA and emerin gene (EMD) were included. Sequencing was performed on the HiSeq 2000 Sequencing platform (Illumina, San Diego, CA, USA) platform. Sequencing data were analyzed using an in-house analysis pipeline. All participants had previously signed an informed-consent form to participate in genetic analysis.
Clinical and histopathologic assessments
Four patients with LMNA-associated muscular dystrophy were included in this study and were confirmed as having an LMNA mutation. The applied variant calling methods were documented previously,9 and validation was performed using Sanger sequencing, for which the concordance rate was 96.3%. We then reviewed the clinical manifestations and findings of neurologic examinations, electromyography, and muscle biopsies from the other hospitals, and compared them with reevaluation findings from our hospital.
RESULTS
Clinical manifestations
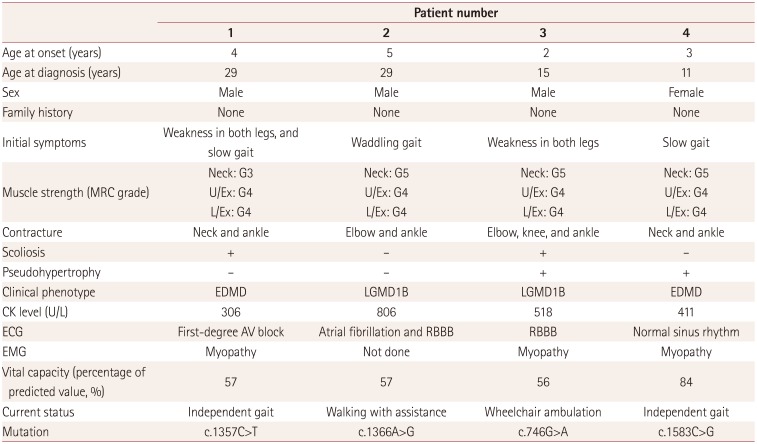
Clinical characteristics and laboratory findings of the subjects are presented in Table 1. Four patients who were confirmed as having LMNA mutations visited other hospitals when their first symptom of slow gait speed or waddling gait occurred between the ages of 2 and 5 years. The presence of contracture was not definitive at the time of the first neurologic examination. Two patients (patients 3 and 4) showed hypertrophy of the gastrocnemius muscles. Sequencing of the dystrophin gene was performed in two patients (patients 2 and 3), who were both negative for mutation in that gene. Without follow-up neurologic examination or evaluation, the patients were admitted to our hospital due to progression of weakness (patient 1 at age 29 years), palpitation (patient 2 at age 29 years), scoliosis surgery (patient 3 at age 15 years), or for re-evaluation (patient 4 at age 11 years). In contrast to their first admission, all of the patients showed prominent proximal muscle weakness and various degrees of distal weakness. All patients showed contracture in the ankle, whereas involvement of the neck extensor was prominent in two patients (patients 1 and 4).
Two patients had scoliosis (patients 1 and 3), and one patient had received an Achilles-tendon-lengthening operation (patient 4). None of the family members had any history of muscle weakness or gait disturbance. The serum creatine kinase level was slightly elevated, ranging between 306 and 806 U/L. Electromyography studies were conducted in three patients (patients 1, 2, and 4), and they showed nonspecific generalized myopathic patterns with low-amplitude, polyphasic motor unit action potentials, and early recruitment patterns. Cardiac evaluation including 24-hour Holter monitoring and echocardiography showed a cardiac conduction block but normal cardiac function in three patients (patients 1, 2, and 3). Three patients had a vital capacity of around 50% of the predicted value, and one of them (patient 3) was using noninvasive positive-pressure ventilation. In one patient (patient 1), CT scans of the lower extremities showed symmetric fatty atrophic changes in proximal and distal leg muscles.
Genetic mutations
We identified a total of four LMNA mutations. The c.1357C>T (patient 1), c.746G>A (patient 3), and c.1583C>G (patient 4) mutations had been previously reported in EDMD patients, while the c.1366A>G mutation found in patient 2 had only been reported in one L-CMD patient.10 Among these mutations, c.746G>A was previously reported in one Korean patient,11 and c.1357C>T has been reported in Chinese patients.12
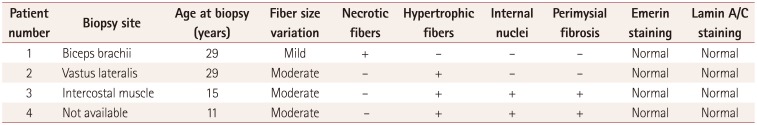
Histopathologic findings
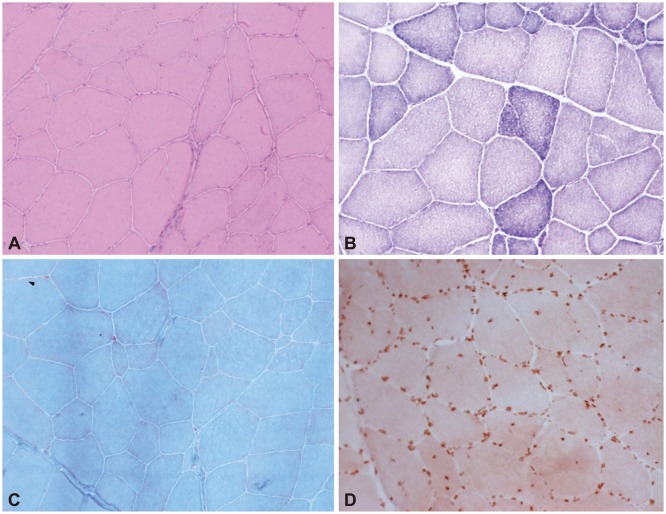
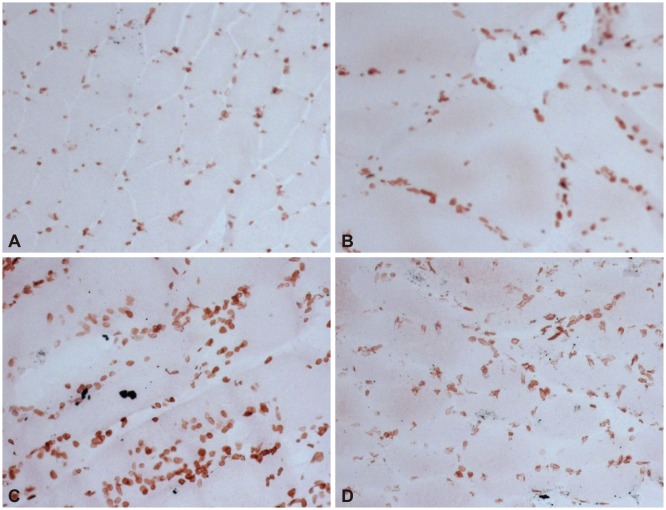
Three patients (patients 1, 2, and 3) had undergone their first muscle biopsy at other hospitals when their first symptoms occurred, but the samples were not available. Since all of the patients had received a muscle biopsy when they were admitted to our hospital, we reviewed their pathologic records from other hospitals and compared them with the findings from the second muscle biopsy at our hospital. The pathologic findings are presented in Table 2. Nonspecific myopathic changes such as size variations in muscle fibers, increased internal nuclei, and hypertrophic fibers with fiber splitting were the main features evident in the patients. Although a few degenerative fibers were observed in patient 1, suggesting muscular dystrophy, they were still nonspecific dystrophic findings (Fig. 1). The findings of immunohistochemistry investigations using antibodies to lamin A/C and emerin were normal compared to healthy controls (Fig. 2).
DISCUSSION
The four patients in this study had clinical phenotypes of EDMD or LGMD1B with known mutations in LMNA. Symptom onset had occurred during early childhood between the ages of 2 and 5 years. The patients had no common features of L-CMD, including infantile onset, delayed development, dropped head syndrome, or inflammatory histopathologic changes.1013 They were initially diagnosed with nonspecific myopathy or other types of muscular dystrophy based on both clinical manifestations and pathologic findings. However, when the patients were admitted to our hospital as adolescents or adults, they showed the typical clinical features of LMNA-associated muscular dystrophy, including progressive proximal muscle weakness with joint contracture and the involvement of cardiac conduction block.
Diagnosing LMNA-associated muscular dystrophy is known to be challenging in the limb-girdle muscular dystrophy phenotype due to the lack of suggestive clinical features. Although the distribution of muscle weakness in patients with LMNA mutations can be divided into limb-girdle and humeropero-neal types, which are phenotypes of LGMD1B and EDMD, respectively, the pattern of muscle involvement often overlaps between the two types. Moreover, early contracture often occurring before muscle weakness is common in EDMD, whereas contracture appears later in LGMD1B and is less severe.
In this study, we found that several clinical factors contributed to the delayed diagnosis of LMNA-associated muscular dystrophy. First, there was later involvement of contracture in early-onset EDMD and LGMD1B patients. It has previously been suggested that later and mild involvement of contracture in LGMD1B contribute to its delayed diagnosis.6 Although early contracture often occurring before muscle weakness is a characteristic feature of EDMD, contracture can appear after weakness with a delay of up to 7 years.3 Although elbow contracture has previously been suggested as an early diagnostic marker,6 we found that most of the patients in our study cohort showed prominent ankle involvement.
Second, patients with calf hypertrophy with prominent proximal weakness were diagnosed as having other types of muscular dystrophy. Calf hypertrophy is known to more often be suggestive of Becker muscular dystrophy and LGMD2I.7 Consortium guidelines published in 1991 suggested the presence of marked calf hypertrophy as an exclusion criterion for X-linked EDMD patients.14 However, as reported previously,3 calf hypertrophy can be observed in laminopathy.
Third, patient education about muscle contracture was not emphasized. A previous study suggested that the involvement of the neck extensor and elbow contracture were the clinical features that were most helpful for an early diagnosis of laminopathy.6 Because muscle weakness is slowly progressive in laminopathy, these patients were not referred to neurologists, and the importance of additional features of muscular dystrophy were not recognized. This resulted in three of the patients receiving tendon-lengthening surgery or a scoliosis operation without visiting neurology clinics.
Three of the four patients underwent muscle biopsies at two different times, with 13 to 25 years between the first and second procedures. Serial histopathologic results from the two biopsies showed nonspecific myopathic changes, and the immunostaining of lamin A/C and emerin appeared normal compared to healthy controls. This shows that serial follow-up muscle biopsies were not informative for the diagnosis in these patients even during the progressive stage of laminopathy. Because of this finding, muscle biopsies are recommended for patients with negative genetic tests.7
The LMNA mutations identified in these patients had been previously reported in EDMD, but the c.1366A>G mutation detected in patient 2 had only been reported once in an LCMD patient. One of the present patients had a clinical phenotype of LGMD1B and reported that symptom onset had occurred at the age of 3 years without dropped head syndrome, which represents the first report of this mutation in an LGMD1B patient.
One study found laminopathy in 4 of 116 congenital muscular dystrophy patients,15 and the application of high-throughput sequencing to patients who were considered to have an unspecified myopathy recently revealed LMNA-associated muscular dystrophy in 5 of 36 patients.12 Even though this is a rare cause of muscular dystrophy, clinicians should consider the possibility of laminopathy and conduct early genetic testing in undiagnosed muscular dystrophy patients with limb-girdle weakness, early humeroperoneal weakness, contracture, or cardiomyopathy due to the possibility of life-threatening cardiac and pulmonary complications.7
 XML Download
XML Download