

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Collagen VI is a ubiquitous extracellular-matrix protein that forms a microfibrillar network, and is closely associated with the basement membrane in most tissues. It is composed of three different peptide chains: α1 and α2 are 140 kDa peptides, while α3 is a 260–300 kDa peptide, and these are encoded by three genes (COL6A1, COL6A2, and COL6A3, respectively).1 Collagen-VI-related myopathy is a disease entity with a broad clinical spectrum, and is caused by mutations in the three collagen-VI-related genes (COL6A1, COL6A2, and COL6A3), resulting in previously identified disorders: Bethlem myopathy (BM; MIM #120250) and Ullrich congenital muscular dystrophy (UCMD; MIM #158810). Both dominant and recessive mutations underlie the entire phenotypic spectrum of these myopathies.
Collagen VI-related myopathy can be classified based on the clinical phenotype into UCMD, intermediate collagen VI-related myopathies (IM), or BM.2 UCMD has been characterized by the congenital onset of muscle weakness, contracture of proximal joints, hyperlaxity of the distal joints, progressive weakness, and respiratory insufficiency.3 Briñas et al.4 referred to patients with UCMD who did not achieve ambulation as “early severe phenotype” and those who did achieve ambulation as “moderate progressive phenotype”. BM typically has a late onset and mild phenotype with a slow or static course, characterized by contractures of ankle and long finger flexors.5 BM was recently described as a myosclerosis variant and collagen VI-related limb-girdle syndrome.2 The IM of collagen VI-related myopathy, called mild UCMD or severe BM, has been identified, but it has not been clearly defined. The genotype–phenotype correlation is very difficult to determine in this myopathy, since the same mutation in the same gene can manifest as diverse clinical symptom severities. The most common mutations in collagen VI-related myopathy are currently missense mutations in the triple-helical domain of COL6A1 and exon-14-skipping with splicing errors in COL6A1.
There have been a few reports on Korean patients with collagen VI-related myopathy. Suh et al.6 reported the clinical and pathologic characteristics in one family with BM. There have also been two case reports of sporadic UCMD with a COL6A1 mutation, as confirmed by a molecular genetic study.78 Chae et al.9 recently reported ten cases of collagen VI-related myopathy (UCMD or BM) with pathogenic or likely pathogenic variants of COL6A1 or COL6A3. Our group has reported one family with BM confirmed by whole exome sequencing and ten cases of collagen VI-related myopathy patients confirmed through targeted next generation sequencing (NGS), but the full clinical information for each patient was not available.1011
To review and analyze the clinical, pathologic, and genetic characteristics of patients with collagen VI-related myopathy, we reviewed data on 22 collagen VI-related myopathy patients including those previously reported by our group. We also explored the genotypes and clinical phenotypes of all collagen-VI-related myopathy patients who had been identified in previous studies.
METHODS
Patient selection, genotype confirmation, and review of clinical information
In our previous study we applied targeted NGS to undiagnosed myopathy patients, which identified ten patients with collagen VI-related myopathy in ten families (families A–J) by targeting 69 genes (Supplementary Table 1 in the online-only Data Supplement).11 We have previously reported five patients in one family (family M) with collagen VI-related myopathy, all of whom were clinically the BM phenotype.10 Collagen VI-related genes were not analyzed in two patients in each of these families (E-02 and M-06), and so we confirmed that those two patients had the same pathogenic mutations as in the other members of their families. In the present study, we added five patients from two families (K-01, K-02, K-03, K-04, and L-01), confirmed by targeted NGS with the same panel used in a previous study.11
We selected 22 collagen VI-related myopathy patients from 13 families in our center who had been reported on previous reports of our group and confirmed by genetic study currently. We reviewed their clinical, pathologic, and genetic features. The clinical information included sex, age, age at onset, physical and neurologic examinations including the joint-contracture site(s), serum creatine kinase level, and results of cardiologic or respiratory evaluations. Based on the clinical presentations in two previous reports,24 we classified our patients into the following three groups:
1) Typical UCMD (moderate progressive phenotype) is classified by early onset, achievement of independent ambulation, loss of walking ability by the age of 20 years, and severe contracture such as scoliosis (patients J-01 and L-01).
2) IM is classified by the ability to ambulate beyond the age of 20 years into early adulthood (patients A-01, F-01, H-01, and I-01).
3) BM is classified by ambulation into adulthood (all of the other patients).
None of our patients presented severe UCMD (early severe phenotype), which is classified by the achievement of sitting only, and sometimes by walking on the knees without independent ambulation along with severe contracture and poor respiratory function.
RESULTS
Clinical and pathologic features of the patients
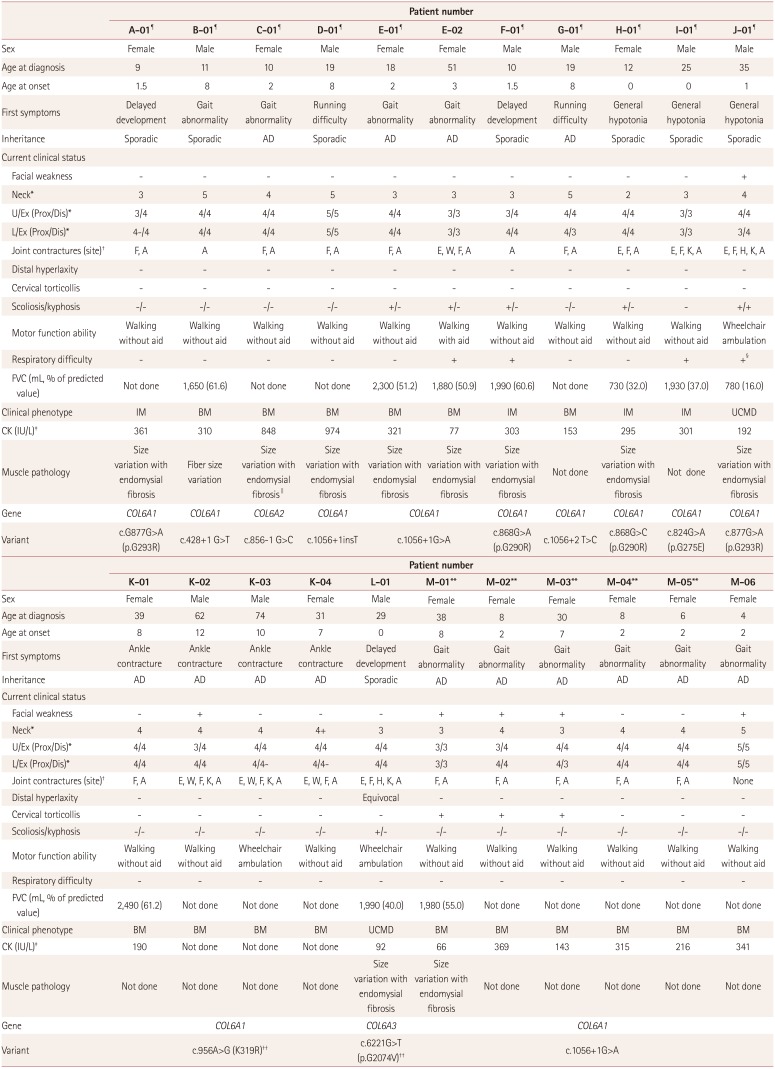
The pedigrees of all patients are presented in Supplementary Figs. 1 and 2 (in the online-only Data Supplement) and we described all information of them in Table 1. Their mean ages at onset and diagnosis were 4.5 and 24.9 years, respectively. The most common initial symptoms were gait abnormality, running difficulty with ankle contracture, and mild lower extremity weakness. Other symptoms included delayed development, general hypotonia, and congenital hip dislocation. Five of the 13 families (families C, E, G, K, and M) showed an autosomal dominant inheritance pattern with a family history, while the others had de novo mutations. Five patients in three families (J-01, K-02, M-01, M-02, and M-03) showed bilateral facial weakness, and no patients showed ptosis or extraocular weakness. Four patients in four families (B-01, D-01, G-01, and M-06) did not show neck weakness or axial muscle weakness, and one of these patients (D-01) did not show limb weakness. All patients except for one 4-year-old girl (M-06) showed joint contracture of the Achilles tendon and/or finger flexors, and three patients (M-01, M-02, and M-03) showed cervical torticollis. Three patients (J-01, K-03, and L-01) needed a wheelchair for ambulation, and one patient (E-02) could walk aided, while the other patients could walk unaided.
The pulmonary function test was applied to 5 of the 16 patients with the BM phenotype, and they showed 50–60% of the predicted value. Only one patient (E-02) suffered from exertional dyspnea and morning headaches. Five of the six patients with IM and the typical UCMD phenotype had forced vital capacities that were less than 60% of the predicted value. Out of these patients, I-01 and J-01 exhibited respiratory difficulties, with patient J-01 only using a ventilator in the supine position due to prominent respiratory difficulties in that position with a low forced vital capacity (16% of the predicted value).
Eleven of the 19 patients who were examined for the serum creatine kinase level showed mild elevation. Electromyography revealed myopathic changes in all of the patients, but they did not exhibit cardiologic abnormalities such as cardiomyopathy or symptomatic arrhythmia (Table 2).
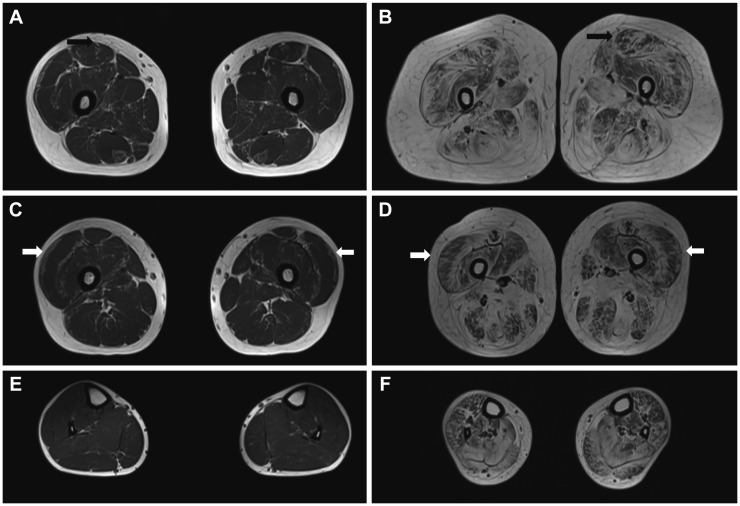
Magnetic resonance imaging of two patients (D-01 and L-01) revealed peripheral fatty infiltration of bilateral thigh muscles (Fig. 1). This finding has been called a “target sign” of the rectus femoris muscle and a “sandwich sign” of the vastus lateralis muscle.12 Clinically we defined patient D-01 as BM and patient L-01 as typical UCMD. Patient D-01 showed mild fat infiltration in the bilateral thigh muscles, while both the thigh and calf muscles of patient L-01 showed moderate-to-severe atrophic changes and fat infiltration.
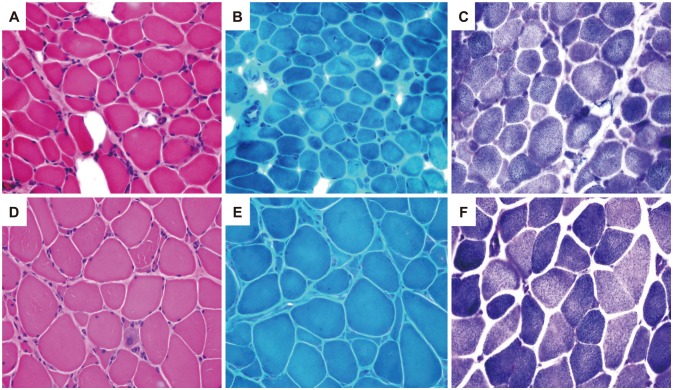
Muscle biopsies performed in ten patients revealed increased muscle fiber size variations with endomysial fibrosis (Fig. 2, muscles from patients A-01 and C-01). The muscle specimen from patient C-01 showed decreased expression of collagen VI in immunohistochemical staining (not shown).
Collagen VI genotypes and correlation with phenotypes
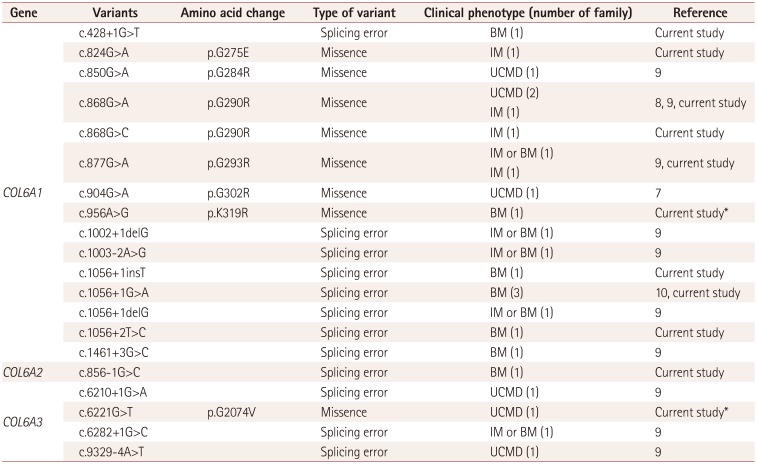
Missense mutations in the triple-helical domain of COL6A1 were the most common genotypes of collagen VI-related myopathy in our series. These mutations were found in patients with both the IM and typical UCMD phenotype. The second most common type of mutations was a splicing-site mutation that resulted in exon-14-skipping. These mutations were found in patients with the BM phenotype and are primarily found in COL6A1, but one mutation each was also found in COL6A2 and COL6A3.
We found five novel mutations in COL6A1 and COL6A2 with clinical phenotypes including those in previous reports (three novel mutations) in our group.11 In the present study, c.956A>G (p.K319R) in COL6A1 resulted in an amino acid change and possible splicing error, while c.6221G>T in COL6A3 resulted in an amino acid change. These variants represented novel mutations that were not found in the public databases (dbSNP 135, Exome Variant Server, and 1,000 Genome Project SNP). We also confirmed that c.956A>G (p.K319R) was not found in normal members of family K, whom we have marked in Supplementary Fig. 1 (in the online-only Data Supplement), and p.K319N was reported to be associated with the UCMD phenotype.13 Meanwhile, c.6221G>A (p.G2074D) in COL6A3, which was at the same site as c.6221G>T, was reported to be associated with a UCMD phenotype.14 Therefore, we could classify these two novel mutations as “likely pathogenic” alleles according to the ACMG/AMP guidelines.15
Our group previously reported three other mutations:11
1) c.856-1G>C in COL6A2 of patient C-01 was near the mutation site of c.856-2A>G, which was previously reported to be associated with the BM phenotype.13 Furthermore, c.856-2A>C was reported in a case with UCMD (rs8860444 66), while c.856-3C>G and c.2386A>G (p.K796X) were reported to be associated with UCMD and in the absence of type VI collagen (from the database of Leiden Muscular Dystrophy; http://www.dmd.nl).
2) c.1056+1insT in COL6A1 was at the same mutation site as c.1056+1G>A, which is a relatively common mutation associated with the BM phenotype.1617
3) The c.428+1G>T variant in COL6A1 was at the same mutation site as c.428+1G>A, and reported to be associated with the BM phenotype.1718
Since all three of these mutations were splicing-site mutations, we could classify them as “pathogenic” alleles according to the ACMG/AMP guidelines.15
We further analyzed the genotype–phenotype correlation in Korea by combining the results of previous studies with the current results (Table 2). The most common mutation was missense mutations in the triple-helical domain of COL6A1. The exon-14-skipping mutation was the second most common mutation, and it was mostly correlated with BM.
DISCUSSION
Collagen VI-related myopathy was previously known to be a two-disease entity comprising BM (mild phenotype) and UCMD (severe phenotype). However, it is now known that these two disease entities are caused by various mutations in the same genes: COL6A1, COL6A2, and COL6A3. Many previous studies have demonstrated the difficulty of distinguishing between the BM and UCMD phenotypes, since there are currently no clear-cut standard boundaries or features for differentiating them. This situation has made it very difficult to establish genotype-phenotype correlations for these two disorders. Foley et al.3 recently reported that pulmonary function profiles could be used in combination with motor function profiles to stratify collagen VI-related myopathy patients in clinical settings.
In this study we investigated the clinical, pathologic, and genetic features of 22 collagen VI-related myopathy patients at our center. There have been some previous reports on collagen VI-related myopathy in Korea: four case reports and one genetic report of ten cases, which included some clinical and pathologic information.678911 In the present study we reviewed the data of 22 patients from 13 families with collagen VI-related myopathy. We analyzed genotype–phenotype correlations not only for our results but also for previous reports involving Koreans. The comprehensive genotype and phenotype review of collagen VI-related myopathy performed in this study is the first of its kind in Korea.
One of the issues focused on this study was defining the clinical phenotypes of collagen VI-related myopathy. We stratified UCMD and IM using a previously reported phenotype stratification method.2419 Based on previous reports, most BM patients present with ambulation into adulthood and ankle contracture with or without mild proximal weakness after 3–4 years of age. Early onset and ambulation status in adulthood could be the main factors for differentiating BM from IM or UCMD. Sixteen patients from 7 of the 13 families with collagen VI-related myopathy in our cohort were defined as BM because they had a relatively late onset (older than 2 years), mild symptoms, and slow progression. Four patients were defined as IM due to early onset (younger than 2 years), delayed development or neonatal generalized hypotonia, walking unaided, and a moderate decrease in the forced vital capacity. Finally, two patients were classified as typical UCMD based on early onset, delayed development or neonatal hypotonia, low forced vital capacity, and current wheelchair ambulation after a period of self-ambulation.
Other than the novel mutations found in the present study, most of the present genotypes have been reported previously for other countries and were similar to the phenotypes of our patients. c.877G>A [family A (IM) and family J (typical UCMD phenotype)] and c.868G>A (family F, IM) in the triple-helical domain of COL6A1 were reported to be associated with various phenotypes, from severe UCMD to the BM phenotype.41617 In addition, c.868G>C (family H, IM) and c.824G>A (family I, IM) in the triple-helical domain of COL6A1 were reported to be associated with mild presentations of UCMD and BM, respectively.1720 c.1056+1G>A (families E and M, BM phenotype) has been reported numerous times, commonly with the BM phenotype and rarely with the UCMD phenotype (database of Leiden Muscular Dystrophy). All of the previously reported cases of this mutation showed the BM phenotype.16172122 In addition, c.1056+2T>C (family G, BM phenotype), which can result in the same exon-14 skipping, is reportedly associated with the BM phenotype.23
BM phenotype was the most common clinical manifestation in the present cohort. Based on previous reports involving Koreans, it is also a frequent clinical phenotype of collagen VI-related myopathy. Furthermore, it is mainly correlated with mutations related to exon-14-skipping,2 with this mutation found less often in the patients with UCMD and IM patients. However, the most common mutation type in the patients with UCMD and IM, including across all of the present patients, is missense mutations in the triple-helical domain. These findings are similar to previous reports from not only Japan and China but also Western countries.21219 Severe UCMD phenotype was not found in the present series of patients, and typical UCMD and IM phenotype were less common than BM phenotype, which is possibly because we mainly see adult myopathy patients at our center. A previous study in Korea found similar prevalence rates of UCMD and BM.9
We have comprehensively reviewed the clinical, pathologic, and genetic features of collagen VI-related myopathy patients in Korea. Although we also reviewed patients included in previous reports, there could be more collagen-VI-related myopathy patients at other centers in Korea, and our data may not represent the actual tendency of clinical phenotypes and genotypes in all Korean patients. It might therefore be useful for a future study to perform a more extensive review of all collagen-VI-related myopathy patients in Korea.
In conclusion, missense mutations in the triple-helical domain of COL6A1 are the most common mutations in collagen-VI-related myopathy in Korea, and patients with these mutations tend to present with an earlier disease onset and more severe progression compared to patients with other mutations. These findings are similar to previous results from Japan, China, and Western countries. We hope that our findings help the understanding of collagen VI-related myopathy and the identification of patients with this disease.
 XML Download
XML Download