

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Nemaline myopathy (NM) is a congenital neuromuscular disease characterized by generalized muscle weakness (predominantly of the respiratory muscles), generalized hypotonia, and the presence of rod-like structures called nemaline bodies in muscle biopsy samples. Mutations are commonly found in the ACTA1 gene.1 We report an NM patient carrying an ACTA1 mutation who also presented with central nervous system (CNS) lesions.
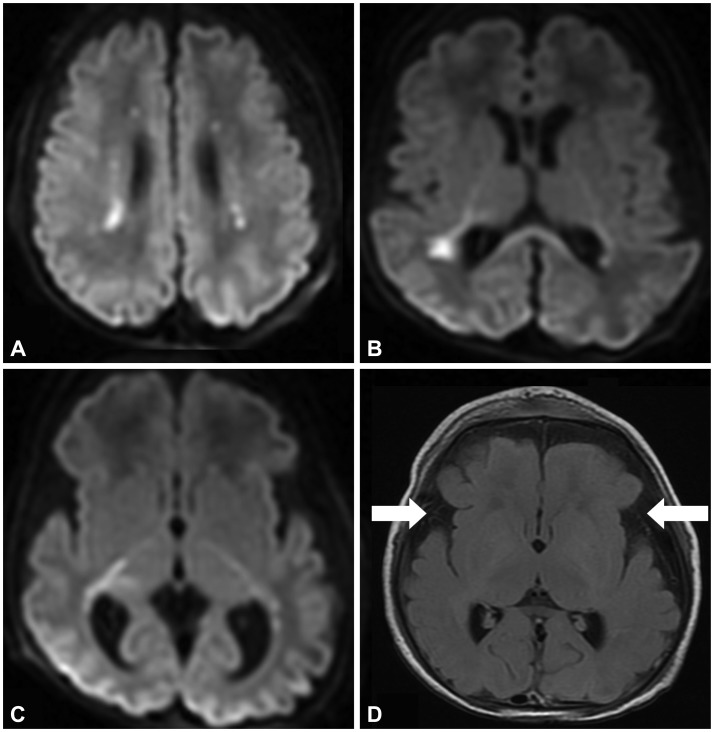
Our patient was a 4-day-old female infant born to nonconsanguineous parents after 35 weeks of gestation in a pregnancy complicated by polyhydramnios. Her family history included the early death (at 2 years) of a maternal half-sister with intrauterine growth retardation and developmental delay. A diagnostic workup including metabolic disease and magnetic resonance imaging (MRI) of the brain did not yield a diagnosis. The patient's weight at birth was between the 75th and 90th percentiles, while her length and head circumference exceeded the 90th percentiles for gestational age. She did not have any dysmorphic features. Immediately after birth she became apneic and required mechanical ventilation. She did not exhibit spontaneous movements in the arms or legs. Her muscle tone was reduced, and deep tendon reflexes could not be elicited. The Apgar score was 1 at 1 minute, 6 at 5 minutes, and 6 at 10 minutes. The cord blood gas showed a pH of 7.36. On day of life 4, MRI of the brain revealed multiple foci of diffusion restriction in the white matter, as well as diffuse prominence of subarachnoid spaces predominantly along the sylvian fissures (Fig. 1). Metabolic disorders were suspected; however, the levels of serum amino acid, urine organic acid, ammonia, lactate, and very-long-chain fatty acid and the acylcarnitine profile were all normal. On day of life 11, intermittent rapid eye movements and lip smacking without alteration of consciousness were noted. Whole-exome sequencing revealed a pathogenic heterozygous, missense mutation (c.868G>C; p.D290H) in the ACTA1 gene on chromosome 1q42.13. Cosegregation analysis of the mutation in this family revealed that the unaffected mother and father did not carry this alteration, indicating a de novo mutation occurrence (Supplementary Fig. 1 in the online-only Data Supplement). The whole-exome sequencing also identified six functionally significant variants in other genes that probably did not contribute to our patient's phenotype (Supplementary Table 1 in the online-only Data Supplement). No potential pathogenic variants or variants of uncertain clinical significance were identified in any myopathy-related genes other than the ACTA1 gene. The patient died on day of life 35 due to sepsis. A postmortem muscle biopsy revealed diffuse myofiber atrophy with many of the fibers containing nemaline rods (Supplementary Fig. 2 in the online-only Data Supplement), which is diagnostic of NM.
NM in the presence of an ACTA1 mutation can present not only with neuromuscular symptoms but also CNS lesions. The ACTA1 gene encodes α-actin, which consists of sarcomeric thin filaments in skeletal muscle and plays a role in cell motility, structure, and integrity. Mutations in the ACTA1 gene cause alterations of the structure or functions of the α-actin, causing the protein to lose its function and aggregate.2 NM is classified into six subtypes based on the severity of skeletal and respiratory muscle dysfunction and the age at onset: severe congenital, Amish, intermediate congenital, typical congenital, childhood onset, and adult onset.3 Patients with severe congenital NM present with severe hypotonia and generalized weakness with respiratory distress and dysphagia at birth. In addition to muscle involvement, CNS involvements including cerebral atrophy, white-matter signal abnormalities, delayed myelination, pachygyria, and diffuse leukomalacia rarely occur in severe congenital and intermediate NM.4
In the present case, MRI revealed CNS lesions, which could be specific to NM. However, the reported pattern of MRI findings can also be observed in other diseases including metabolic disorders, infectious diseases, or hypoxic ischemic encephalopathy (HIE). Metabolic and infectious etiologies were less likely because of the normal metabolic workup and the lack of evidence of infection. Despite the unusual pattern of the white-matter injury and the favorable pH for HIE, a hypoxic ischemic insult was a possibility. We often encounter patients who are suspected of having metabolic disorders or HIE but where the exact etiology remains unknown. Since NM can present with CNS lesions mimicking other disorders, it may be useful to include NM in the differential diagnosis of neurological disorders with hypotonia and abnormal brain MRI findings.
 XML Download
XML Download