

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hypertrophic pachymeningitis (HP) is a rare disorder showing chronic fibrous inflammation of the dural meninx.1 HP causes various neurologic symptoms associated with dural thickening, since compression near the parenchyma can induce seizure, paralysis, and ataxia. Sinus drainage is hindered, resulting in cerebral edema and venous infarction. Direct invasion of the cranial nerves leads to multiple cranial nerve palsies, and irritation of the dura mater itself generates headache and nausea. Inflammation often spreads to adjacent structures, causing mastoiditis and sinusitis.1234
The exact mechanism underlying HP is still unknown, but speculative etiologies include an external insult, such as a direct infection by a virus or fungus, or systemic autoimmune diseases such as systemic lupus erythematosus, Wegener's granulomatosis, or systemic IgG4-related disease.56 Several cases are classified as idiopathic HP, in which the pathologic findings are lymphoplasmacytic infiltrates with fibrous proliferation.7
The treatment of HP is based on the possible underlying autoimmune mechanism. Corticosteroid is conventionally recommended as the first-line therapy for HP, but its long-term benefits are limited, and a refractory fate is common following the cessation of therapy. Rituximab (RTX), a CD20 selective inhibitor, was reported to be an effective second-line therapy in steroid-refractory patients with IgG4-related disease and Wegener's granulomatosis.89 Given the close relation of these diseases with HP, we identified three patients with steroid-refractory idiopathic HP who reached complete remission with RTX treatment. We suggest that RTX is a good alternative therapy for steroid-refractory HP, even for cases with idiopathic etiologies. The use of RTX and the analysis of the patients in this study were approved by the Institutional Review Board of Seoul National University Hospital.
METHODS
Patient 1
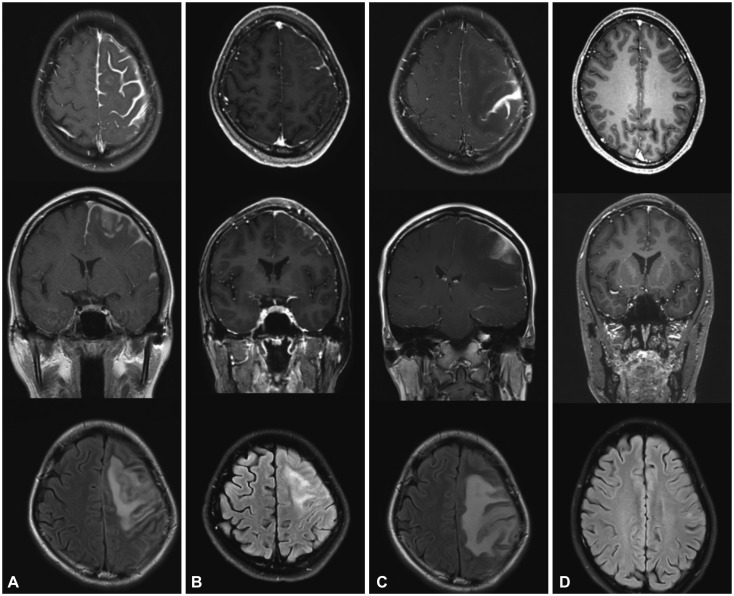
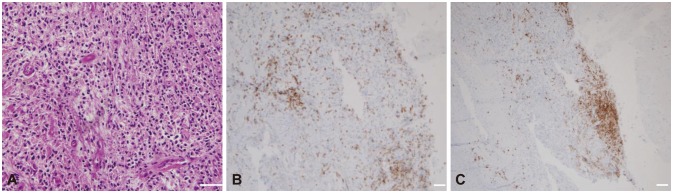
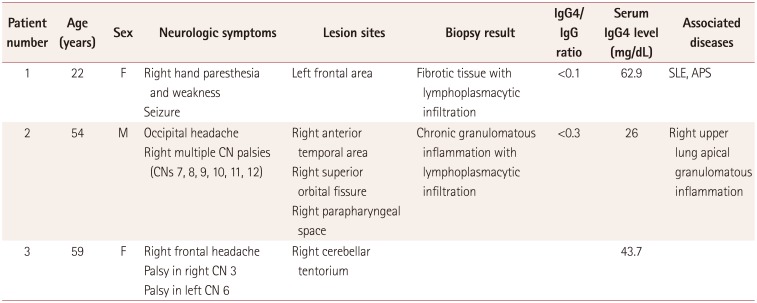
A 22-year-old woman developed paresthesia in the distal extremities. The condition began insidiously, and the patient experienced a transient severe tingling sensation on the right arm for 2 hours on the day prior to her emergency room (ER) visit. She presented at the ER with generalized clonic seizure, and MRI revealed pachymeningeal and leptomeningeal enhancement in the left frontal area (Fig. 1A). A brain biopsy showed fibrotic tissue with lymphoplasmacytic infiltration, showing focal positivity of IgG4 immunostaining (IgG4/IgG ratio <0.1) consistent with pachymeningitis (Fig. 2).
The patient was also suspected as having a systemic autoimmune disease based on the appearance of autoimmune antibodies, but no disease fulfilled the criteria. She was positive for antinuclear, anti-Smith, and anticardiolipin antibodies, at titers of 1:320, 10.2, and 46.1, respectively. Her anti-double-strand DNA antibody and serum IgG4 levels were within the normal ranges.
Along with anticonvulsant (levetiracetam) therapy, she was treated with prednisolone (3 weeks, 40 mg/day), azathioprine (3 months, 25 mg/day), and hydroxychloroquine (maintained, 100–200 mg bid). The steroid was maintained at a low dose (5 mg/day), but azathioprine was held due to a low absolute neutrophil count level (835 neutrophils/mm3). During the first month of treatment, MRI showed improvement of the meningeal enhancement and cortical swelling (Fig. 1B).
After 7 months of treatment, the patient complained of right-hand clumsiness. MRI revealed aggravation in the left frontal parietal area (Fig. 1C). Along with prednisolone (60 mg/day) again for 1 week, RTX therapy (375 mg/m2) was administered for four cycles. The patient's paresthesia and clumsiness improved during the second and third cycles of RTX. A 3-month serial follow-up with brain MRI revealed a gradual improvement of pachymeningitis, including less T1-weighted enhancement at the meninx and parenchyma (Fig. 1D). The patient exhibited no recurrence of any neurologic symptoms during an 8-month follow-up after RTX treatment.
Patient 2
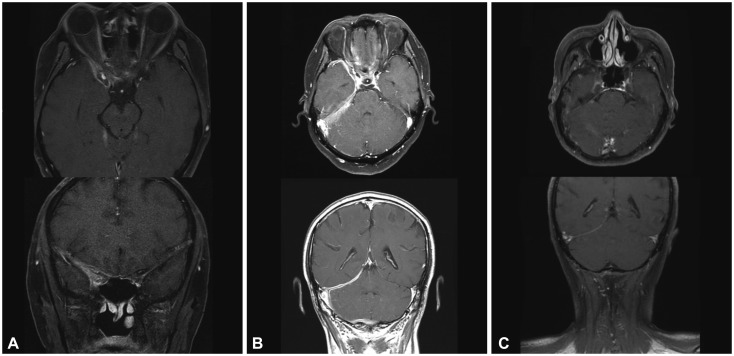
A 54-year-old man suffered a severe occipital headache with multiple cranial nerve palsies on the right side, manifesting as hoarseness, dysphagia, uvular deviation, tongue deviation, and shoulder elevation weakness. Brain T1-weighted MRI with gadolinium revealed an ill-defined enhancement along the right anterior temporal area, superior orbital fissure, and parapharyngeal space (Fig. 3A). A biopsy at the right Rosenmüller fossa demonstrated chronic inflammation with plasma cell infiltration, showing focal positivity in IgG4 immunostaining (IgG4/IgG ratio<0.3) (Fig. 4).
PET-CT revealed hypermetabolic lesions not only in the right retropharyngeal area but also in the right upper lung apex. A percutaneous needle biopsy at the multiple pulmonary lesions showed chronic granulomatous inflammation with necrosis, presenting normal results for acid-fast bacilli staining, tuberculosis PCR, and IgG4 immunostaining.
Laboratory findings for bacterial, viral, tuberculosis, and fungal infections were all negative in the serum and cerebrospinal fluid (CSF). The serum levels of antinuclear antibody, antineutrophil cytoplasmic antibody, and IgG4 were within the normal ranges.
The patient received steroid therapy (prednisolone at 50 mg/day orally), and showed a good initial response in both the brain and lung. However, the steroid could not be tapered due to the wax-and-wane pattern of his neuronal symptoms. Azathioprine was added, but the patient still complained of headache.
Despite applying immunotherapy for 7 months, follow-up MRI showed aggravated thickening of the dura mater (Fig. 3B). A repeated biopsy at the right superior orbital fissure showed chronic granulomatous inflammation consistent with idiopathic pachymeningitis.
The patient was administered RTX therapy (375 mg/m2) for four cycles. After 4 months of RTX treatment, the patient's symptoms disappeared, and brain MRI revealed decreased thickening of the dura (Fig. 3C). Following four additional cycles of RTX for maintenance, the patient was symptom-free during the 23-month follow-up.
Patient 3
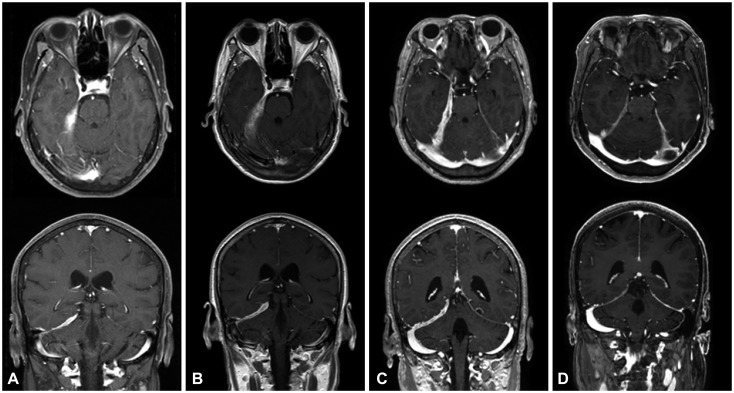
A 59-year-old woman presented with binocular diplopia and headache in the right frontal area. A neurologic examination revealed that the patient had impaired medial gaze in the right eye without pupil dysfunction or ptosis, implying right oculomotor nerve palsy. Brain MRI showed dural thickening along the right cerebellar tentorium (Fig. 5A). In accordance with the protocol for idiopathic pachymeningitis, the patient was administered steroid therapy (prednisolone at 1 mg/kg/day orally) for 1 week. The steroid was tapered as the symptoms improved.
However, the patient complained of diplopia 1 month later. She presented with lateral gaze limitation of the left eye consistent with left abducens nerve palsy. Brain MRI revealed an increased extent of dural thickening compared with the previous MRI investigation (Fig. 5B). Steroid pulse therapy (methylprednisolone at 1,000 mg/day) was applied for 5 days, which resulted in disappearance of the patient's symptom.
Diplopia recurred for the third time 1 month later, with left oculomotor nerve palsy. Brain MRI again revealed an increased extent of dural thickening (Fig. 5C). A CSF study showed mild leukocytosis (26 white blood cells/mL) with elevated protein (115 mg/dL). No microorganisms were detected in the patient's blood or CSF. The laboratory results for autoimmune antibodies were normal, including for IgG4, anti-neutrophil cytoplasmic antibody (ANCA) (myeloperoxidase), ANCA (proteinase III), and angiotensin-converting enzyme.
Four cycles of RTX therapy (375 mg/m2) were administered for the patient's refractory idiopathic pachymeningitis. The patient experienced no symptoms for 12 months up to the last follow-up in May 2016, consistent with decreased thickening of the dura mater found in follow-up brain MRI performed after 6 months of RTX treatment (Fig. 5D).
RESULT
Case summary and analysis
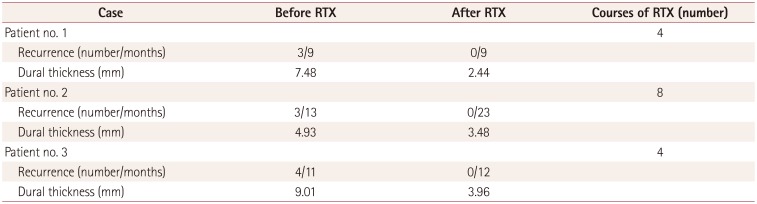
The clinical responses were assessed based on the number of recurring neurologic symptoms during the period from the first attack of HP to RTX treatment and from starting RTX to the last follow-up in May 2016 (Table 1). The dura mater appeared thickest in gadolinium-enhancement MRI. All of the patients became free of the recurrence of neurologic symptoms after the RTX therapy. The dura mater thickness in the three patients decreased from 7.48 mm to 2.44 mm, from 4.93 mm to 3.48 mm, and from 9.01 mm to 3.96 mm (Table 2).
DISCUSSION
RTX was effective as a second-line therapy for steroid-refractory idiopathic HP in the three cases reported here. RTX is a well-known secondary option for systemic IgG4-related disease and Wegener's disease, and there are some case reports of the effectiveness of this drug when the aforementioned diseases are combined with HP.91011 To the best of our knowledge, this is the first article on RTX being applied to idiopathic HP with the results suggesting RTX as a good rescue option for HP cases.
Nonspecific chronic inflammation of the dura mater is a common neuropathologic feature of idiopathic HP. Lymphoplasmacytic cells, histiocytes, and epithelioid cells infiltrate the thickened dura mater, subsequently being replaced by fibrosis. Vasculitis and granulomatous changes are seen occasionally.7 Active inflammatory areas are responsive to corticosteroid as the first-line treatment of idiopathic HP.5 However, remission is often not achieved with a corticosteroid, with one report of a relapse rate of 46%.12 Alternative immunosuppressive agents such as azathioprine, cyclophosphamide, and methotrexate have been tested in patients who were refractory to steroids or developed steroid dependence, and these agents showed long-term success in a few patients.121314
RTX is a chimeric monoclonal antibody that binds to CD20 protein present on the surface of pan-B cells and selectively suppresses B-cell-involved autoimmunity.15 This has resulted in the increasing use of RTX in autoimmune diseases, replacing corticosteroids and other cytotoxic immunotherapies. IgG4-related disorder (IgG4 RD) is a newly recognized autoinflammatory condition induced by the overactivity of T2-helper cells, stimulating plasma cells to produce excessive IgG4. Fibroinflammation creates tumefactive lesions in virtually every organ system.16 IgG4 RD occasionally invades the meninges, causing HP. A recent paper reported that 66% of previously diagnosed cases of idiopathic HP were actually IgG4 RD.6 Treatments for IgG4 RD target hyperactive B cells with RTX, suppressing the reciprocal activation of T2-helper cells to relieve the systemic inflammation level.10
Two of the present patients in whom a biopsy was performed showed no evidence of IgG4 RD, and the serum IgG4 levels of all patients were within the normal range. No criteria of other autoimmune diseases were fulfilled, and their HP was therefore diagnosed as idiopathic. Because lymphoplasmacytic infiltration was demonstrated in the pathology of two of the patients, B-cell hyperactivity might play a crucial role in the pathomechanism of idiopathic HP, as it does in IgG4 RD.6 This supports the use of RTX as a second-line treatment, resulting in long-term complete remission. This success has additional meaning in that two of the present patients had previously failed to maintain a combination therapy with azathioprine (a conventional alternative) due to side effects and the occurrence of relapse.
In conclusion, this study has demonstrated that RTX is a feasible option for HP treatment with long-term remission. However, because only three patients were included, an additional study involving a large population is required to establish RTX as a guideline treatment. In addition, given the good efficacy in the reported cases, RTX can be considered as a first-line treatment in HP, as it is in ANCA-associated granulomatous disease.17

 XML Download
XML Download