

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
22q11.2 deletion syndrome (22q11.2DS) is the most frequent interstitial deletion syndrome in humans, and is caused by a 3-Mb hemizygous DNA microdeletion encompassing approximately 60 known genes on the long arm of chromosome 22.1 The prevalence of 22q11.2DS is estimated to be 1 in 4,000 live births, and 90% of these mutations arise de novo.2 More than 150 highly variable phenotypes of 22q11.2DS can manifest in multiple organs and tissues. Characteristic symptoms include congenital heart disease (CHD), palatal abnormalities, hypoparathyroidism, facial dysmorphisms, velopharyngeal insufficiency, T-cell abnormalities, mild-to-moderate cognitive deficits, and psychiatric illness. This phenotypic variability is considered to be related to breakpoint heterogeneity and other familial and environmental factors.3
Previous studies have found diverse neuropsychiatric (NP) manifestations to be the primary phenotype of this genetic syndrome, and they have suggested that several genes located within the deleted region are linked to these phenotypes.45 The NP manifestations in 22q11.2DS are diverse, including developmental delay, cognitive impairment, behavioral or psychiatric issues, and epilepsy,2 and they significantly affect the quality of life. Structural central nervous system (CNS) abnormalities including polymicrogyria, cerebral atrophy, and cerebellar hypoplasia have also been reported as primary manifestations or coincidental associations.467 However, NP manifestations in 22q11.2DS cases other than cognitive impairment and psychiatric disorders are still not well-characterized. Despite their high prevalence and various clinical symptoms, NP manifestations of this genetic syndrome can be underestimated due to a late appearance and relatively mild presentation.8910
Among the NP manifestations of 22q11.2DS, seizures are conspicuous events that result in neurological consultations, and epilepsy is a chronic disease that requires long-term follow-up in the neurology clinic. The disproportionate associations between 22q11.2DS and generalized epilepsy or juvenile myoclonic epilepsy1112 suggest that this microdeletion plays a role in genetic generalized epilepsy. Moreover, polymicrogyria and other malformations of cortical development,1314 which are major causes of epilepsy, are relatively common in 22q11.2DS.71516 However, the role of 22q11.2DS in epilepsy and brain development is still unclear.
To delineate the relationship between 22q11.2DS and the development of epilepsy, the present study aimed to characterize epilepsy in children with 22q11.2DS and to identify the coexisting NP phenotypes in 22q11.2DS and epilepsy. In addition, we evaluated a potential link between epilepsy and other NP phenotypes in this genetic neurodevelopmental disorder.
METHODS
Subjects
This study included 145 children and adolescents with 22q11.2DS who presented at the Asan Medical Center Children's Hospital between 1996 and 2013. The diagnosis of 22q11.2DS was confirmed in all participants using fluorescence in situ hybridization or multiplex ligation-dependent probe amplification. Data on sex, age, ages at presentation and diagnosis, family history, birth-related history (gestational age and birthweight), initial manifestations, growth profiles, and other characteristic phenotypes associated with 22q11.2DS were obtained by a retrospective review of the medical records. The initial manifestations and presenting phenotypes associated with 22q11.2DS involved facial dysmorphisms, growth retardation, congenital heart defects, velopharyngeal abnormalities, parathyroid dysfunction, immune deficiency, microcephaly, and CNS manifestations including epilepsy, developmental delay, and psychiatric illness. Epilepsy was classified etiologically according to the 2010 International League Against Epilepsy classification.17 Epilepsy without a distinct structural or metabolic condition was defined as genetic epilepsy in the present 22q11.2DS patients, where the epilepsy is understood to directly result from a known or presumed genetic defect. Structural epilepsy in 22q11.2DS was defined when there were distinct structural abnormalities associated with a substantial risk of developing epilepsy. Developmental delay was defined as a failure to reach appropriate developmental age-related milestones in motor skills and cognitive function. Psychiatric disorders were clinically diagnosed by a pediatric psychiatrist (H.W. Kim) based on the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. In patients with reported epilepsy and other NP symptoms, detailed neurological examinations, electroencephalogram (EEG) recording, brain imaging studies (ultrasonography, computed tomography, or magnetic resonance imaging), and psychological examinations were performed according to their symptoms. Clinical variables were also reviewed, including age at the onset of seizures, seizure type, antiepileptic drugs used, comorbidities of other NP manifestations, and laboratory data. The overall and individual prevalence rates of the various NP phenotypes were analyzed, and the patients with epilepsy were categorized into two groups based on etiology and described in detail. To investigate the possible associations between epilepsy and other clinical factors, the clinical factors were compared between 22q11.2DS patients with and without epilepsy.
This study was approved by the Institutional Review Board of the Asan Medical Center, University of Ulsan College of Medicine, Korea. Informed consent was provided by the parents or guardians of the study subjects.
Data analysis
Clinical variables were compared across 22q11.2DS patients with and without epilepsy using independent t-tests for the continuous variables and χ2 and Fisher's exact tests for the categorical variables. All statistical analyses were performed using SPSS statistical software (version 18.0, SPSS, Chicago, IL, USA). A p value of <0.05 was considered indicative of statistical significance.
RESULTS
Clinical and NP manifestations in 22q11.2DS
One hundred and forty-five patients (72 males and 73 females) with a diagnosis of 22q11.2DS were included. The median patient age at enrollment was 6.6 years [interquartile range (IQR)=2.5-12.1 years], and the median duration of follow-up was 3.9 years (IQR=0.8-9.1 years). The median age at diagnosis was 10.3 months (IQR=1-47 months), and a prenatal diagnosis was made in one case. The most common initial manifestation at diagnosis was CHD (n=105, 72.4%), followed by NP symptoms of developmental delay (n=13, 9.0%) and epileptic seizures (n=2, 1.4%). Fig. 1A describes the presenting phenotypes and their frequency at the time of study enrollment. NP disorders (n=66, 45.5%) were the third most common manifestation (Fig. 1A), and 25 (37.9%) among those 66 patients showed at least two coexisting NP symptoms (Fig. 1B). The most common NP manifestation of 22q11.2DS was developmental delay or intellectual disability (n=58). Other common psychiatric disorders were attention-deficit/hyperactivity disorder (ADHD) (n=9) and mood disorders (n=4) (Fig. 1B).
Epilepsy in 22q11.2DS
Twenty-two of the patients (15.2%) presented with epileptic seizures. The median age at seizure onset was 8 months (IQR=4-33), and 16 patients experienced their first seizures when they were younger than 2 years. The most common type of seizure was generalized tonic-clonic seizure (n=16, status epilepticus in one) followed by focal clonic seizure (n=4) and myoclonic seizure (n=2). Among the 22 patients with epileptic seizures, 12 patients (8.3%; 5 males and 7 females) were diagnosed as having genetic epilepsy (Table 1). The types of seizures in the genetic epilepsy cases were mostly generalized seizures: the exceptions were one patient with focal clonic seizures and a second with subtle seizures with desaturation. EEGs were obtained in the 12 patients with genetic epilepsy (Table 1), of which 6 (50.0%) had a developmental delay or intellectual disability and 2 (16.7%) had psychiatric disorders: 1 with ADHD and mood disorder and 1 with a mood disorder.
Epilepsy with a structural etiology was diagnosed in 10 patients (Table 2). The type of seizures and EEG findings are described in Table 2. Among these 10 patients with structural epilepsy, 9 patients (90.0%) had developmental delay or intellectual disability and 3 (33.3%) had comorbid psychiatric disorders, comprising autism spectrum disorder (ASD), ADHD, and mood disorder. Developmental delay was significantly more common in the 22q11.2DS patients with epilepsy than in those without epilepsy (p=0.003) (Table 3). The risk of epilepsy increased significantly when a 22q11.2DS patient showed a developmental delay, with an odds ratio of 3.98 (95% confidence interval=1.5-10.5; p=0.005). A psychiatric disorder was also more frequent in patients with an epilepsy phenotype (5/22, 22.7%) than in those without epilepsy (11/123, 8.9%), but there was no statistical significance (p=0.057). In comparing the etiologies of epilepsy, developmental delay was found to be significantly associated with structural epilepsy (p=0.045), whereas a psychiatric manifestation did not show this relationship.
Structural brain abnormalities in 22q11.2DS
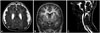
There were abnormal findings in 18 of the 53 patients (34.0%) who underwent brain imaging studies, which revealed congenital CNS abnormalities in six patients. Malformations of cortical development, including diffuse polymicrogyria (n=3) (Fig. 2A) or focal cortical dysplasia and subependymal heterotopia (n=1) (Fig. 2B), were demonstrated in four patients, and Chiari malformation was present in one patient (Fig. 2C) and hypoplasia of the intracranial artery in another patient. Various acquired CNS abnormalities were observed, including a cerebral infarction (n=4), diffuse or focal atrophy (n=3), cerebral hemorrhage (n=2), calcification (n=2), and multifocal white-matter changes (n=1).
Comparison of clinical variables between 22q11.2DS patients with and without epilepsy
We examined the possibility that other clinical variables affected the occurrence of the epilepsy phenotype in the current patient series (Supplementary Table 1 in the online-only Data Supplement). Comparing sex, age at diagnosis, microcephaly, facial dysmorphism, hypocalcemia, severity of CHD, and birth-related factors between 22q11.2DS patients with and without epilepsy revealed that none of these variables was associated with the development of the epilepsy phenotype in this genetic syndrome.
DISCUSSION
Microdeletion syndromes such as 22q11.2DS present extremely heterogeneous phenotypes and different neurodevelopmental disorders, and 22q11.2DS is a prototypic microdeletion syndrome.18 Previous studies have already shown that NP manifestations are frequent in 22q11.2DS.411 However, few studies have focused on epilepsy in this syndrome, and the role of 22q11.2DS in epileptogenesis is still unclear. The current study focused on other overlapping NP manifestations and the electroclinical features of epilepsy as well as the overall incidence of epilepsy in 22q11.2DS patients, and found that epilepsy is a common phenotype for 22q11.2DS and that cognitive impairment or psychiatric disorders also significantly coexist in 22q11.2DS. These findings suggest that this specific genetic locus is critically linked to epileptogenesis and neurodevelopment.
Similar to previously reported findings,219 the most common NP manifestations of 22q11.2DS in the current cohort were developmental delay and intellectual disability. Although the current study was mostly based on parental or caregiver reports of developmental milestones and school performances, this result is in agreement with several previous studies using neuropsychological tests89 to find psychomotor and language developmental delay, or specific deficits of numerical and visuospatial performance, in 30-40% of 22q11.2DS patients. In addition, variable psychiatric disorders were diagnosed in 11.0% of the patients in the present study. Previous research has shown high incidence rates of anxiety disorders, schizophrenia spectrum disorders, ADHD, mood disorders, ASD, and oppositional defiant disorder in 22q11.2DS.1920 However, the relatively low incidence rates of mood disorder and schizophrenia in the current study could have been due to the youngness of the subjects at enrollment and the short follow-up duration during adolescence or adulthood.
The current analyses revealed that 22q11.2DS patients have an increased risk of both provoked and unprovoked seizures, which is consistent with the findings of previous studies.1121 The prevalence of epilepsy in the present cohort was 15.2%. Excluding patients with structural CNS abnormalities that contribute to the occurrence of seizure, the prevalence of genetic epilepsy was 8.3%, which is much higher than frequencies of primary epilepsy in 22q11.2DS of 3.7% and 4.8% found in two previous studies.1121 The prevalence of genetic epilepsy in the present study might have been overestimated, and there can be overlap between structural and genetic epilepsy in this particular syndrome. However, several electroclinical features of epilepsy in 22q11.2DS provide strong evidence of a link between genetic epilepsy and 22q11.2DS. Most of our patients with genetic epilepsy presented with generalized seizures, including myoclonic ones, which is compatible with a previous study finding an association between juvenile myoclonic epilepsy and 22q11.2DS.1222 The clinical generalized seizures identified in the present analyses were also found to be associated with generalized EEG abnormalities, in agreement with previous findings,11 which is not specific to 22q11.2DS but suggests an association with genetic generalized epilepsy. Microdeletion syndromes including 22q11.2DS are significantly related to genetic generalized epilepsy with intellectual disability,182324 and, conversely, patients with epilepsy frequently have large copynumber variations when they exhibit mental retardation or other NP problems.24 A consistent finding is that patients with epilepsy also have a higher risk of developmental delay and mental retardation than those without epilepsy, since this indicates a close relationship between epilepsy and other neurodevelopmental disorders.
The findings of the present study also demonstrate the variability of CNS abnormalities-from a minimal change in the white matter to malformations of cortical development-in patients with epilepsy, as reported previously.467 Among the detected structural abnormalities, malformations of cortical development including polymicrogyria, focal cortical dysplasia, and subependymal heterotopia appeared relatively frequently in the current patient series, as found in previous studies.1314 There are several recent reports of microdysgenesis of the brain in genetic generalized epilepsy or juvenile myoclonic epilepsy.2526 As indicated above, epilepsy that arises in 22q11.2DS is closely associated with genetic generalized epilepsy, suggesting the presence of microscopic morphological changes in the brain. These gross and microscopic morphological variations should be considered in parallel with the heterogeneous electroclinical features of epilepsy.
An association of psychiatric morbidity in 22q11.2DS with morphological alteration of the brain-including a reduced volume of the gray matter in the superior temporal lobe, enlarged Sylvian fissures, and posterior fossa reduction-has also been described previously.27 Furthermore, the present study also demonstrated other rare congenital brain anomalies related to the syndrome such as an anomaly of the cerebral vasculature like hypoplasia of the intracranial artery21 and a Chiari malformation.28 Various imaging and histopathological findings in 22q11.2DS have been attributed to a direct effect of the chromosomal microdeletion,14212223 and active surveillance for brain malformations can help patients with 22q11.2DS and NP manifestations. The disrupted expression of multiple genes involved in the development and maturation of neurons, neurotransmission, and neuronal circuits45 can explain the broad spectrum of structural abnormalities, epilepsy, and the coexistence of several NP manifestations in 22q11.2DS.
Given that some patients with epilepsy have acquired structural etiologies, there is the possibility of developing epilepsy secondarily via severe CHD with hypoperfusion, antiepileptic drug medication, or environmental factors. Previous studies have found that other congenital anomalies of 22q11.2DS can impact early brain development and the NP phenotype from infancy to adolescence.4 In particular, recurrent and chronic cerebral hypoperfusion caused by CHD is suggested to influence the alteration of early neurodevelopment and the high incidence of psychiatric disorders.7 In contrast, other recent studies have demonstrated that the presence of CHD in 22q11.2DS does not have a major impact on neurocognitive deficits and the incidence of psychiatric disorders,2930 and the present findings also confirm that microcephaly, characteristic facial features, the severity of CHD, and birth-related factors are not major contributors to epilepsy in 22q11.2DS.
The current study was limited by its retrospective design, insufficient data about the onset of NP manifestations, and the lack of comprehensive neuropsychological examinations. There was also the possibility of underdiagnosis in patients with belatedly appearing or atypical phenotypes. Additionally, data regarding the exact breakpoints of the 22q11.2DS patients were not available in this study. The relatively small number of patients who underwent brain imaging studies or EEG recording is another limitation.
In conclusion, there are diverse and coexisting presentations of epilepsy and other NP phenotypes in children and adolescents with 22q11.2DS. Diagnostic screening and early targeted intervention for their NP manifestations is important to improving patient outcomes. Future studies investigating the neurobiological basis of NP manifestation using advanced genetic technologies and functional studies will further promote our understanding of epilepsy and other NP phenotypes in 22q11.2DS.




 XML Download
XML Download