

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Myotonia congenita (MC) is a group of genetically and clinically heterogeneous congenital neuromuscular channelopathies characterized by the delayed relaxation of the muscles after voluntary contraction, stiffness, hypertrophy, transient weakness, and cramping. MC is mostly associated with dominant or recessive mutations in CLCN1. However, SCN4A mutations are occasionally implicated in dominant MC. Besides MC, SCN4A mutations are reportedly associated with many other allelic disorders with overlapping phenotypes, including paramyotonia congenita (PMC), hyperkalemic/hypokalemic periodic paralysis, and congenital myasthenic syndrome.123 Moreover, several mutant alleles of SCN4A exhibit a wide intra-allelic phenotypic spectrum (e.g., p.S804F, p.A1156T, p.G1306V, and p.V1589M).45 The majority of hypokalemic periodic paralysis cases are caused by mutations in CACNA1S, the gene encoding the skeletal muscle voltage-gated calcium channel α-subunit.6
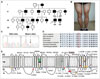
This study examined a 19-year-old man who had experienced painless stiffness of the hands since the childhood (Fig. 1A, III-9). His muscle stiffness was worse on the initiation of exercise and aggravated by severe cold, and reduced by repetitive contraction. He had never been able to play any sport. Weakness or episodic paralysis did not occur. Twelve family members including his father and brother had experienced the same symptoms. An examination revealed myotonia in all four limbs. Motor weakness and percussion myotonia were absent, and tendon reflexes were normal. His muscles were very well developed, especially in the lower limbs (Fig. 1B). The serum electrolyte and creatine kinase levels were normal. There was no abnormality in nerve conduction, but needle electromyography revealed diffuse continuous myotonic discharges that were accentuated by needle displacement with dive-bomber sounds. Short-duration exercise induced transient decreases in the amplitudes of the compound muscle action potentials (CMAPs) immediately after the exercise. Performing the short-duration exercise at cold also caused postexercise reduction in CMAP amplitude. These decreases in CMAP amplitude were larger when the exercise was performed at a lower temperature (Supplementary Fig. 1 in the online-only Data Supplement).
Exome sequencing revealed a c.G3877A (p.V1293I) mutation in SCN4A. The mutation was co-segregated with the affected members within the family (Fig. 1A and C). The p.V1293I mutation was observed neither in 302 controls nor in several global human variant databases, such as the 1000 Genomes Project and the Exome Sequencing Project (Supplementary Table 1 in the online-only Data Supplement). The mutation was located in the highly conserved transmembrane domain (Fig. 1D), and in silico predictions also suggested a pathogenic effect (SIFT: 0.00, PolyPhen2: 0.765, and MUpro: -0.942). These findings imply that the p.V1293I mutation is the underlying cause of the MC symptoms. Other nonsynonymous variants observed in myotonia-related genes were considered to be benign variants, because they were polymorphic or nonsegregated with the affected individuals (Supplementary Table 1 in the online-only Data Supplement).
The p.V1293I mutation was previously reported in a German PMC family with characteristics that differed from our MC case.2 Therefore, the present report may be of the first case of p.V1293I mutation in SCN4A as the underlying cause of MC. As shown in Fig. 1E, SCN4A mutations have revealed wide inter- and intra-allelic clinical heterogeneities.
This study suggests the presence of a phenotypic variability of the p.V1293I allele with overlapping features of MC and PMC. It also suggests that patients with nondystrophic myotonia should be screened for mutations in SCN4A in addition to CLCN1.
 XML Download
XML Download