

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Central core disease (CCD, MIM #117000) is a rare congenital myopathy that is generally inherited as an autosomal-dominant (AD) trait, although there are a few reports of autosomal-recessive or sporadic cases.1,2,3 It is allelic to malignant hyperthermia caused by mutation in the gene encoding ryanodine receptor 1 (RYR1), and is triggered by inhalational anesthetics and depolarizing muscle relaxants.1,4 The severity of the clinical features is highly variable, ranging from severe neonatal disease to mild symptoms. Most patients have slowly or nonprogressive proximal muscle weakness, muscular hypotonia during infancy, and delayed motor development.5 Most of the affected individuals have involvement of the facial and neck muscles and musculoskeletal alterations including hip dislocation, joint contracture, and kyphoscoliosis.5,6 Histochemical staining of muscle samples reveals the characteristic absence of oxidative enzyme activity in core regions.7 Most cases with clinicopathological features of CCD are associated with mutations in the skeletal muscle gene RYR1 on chromosome 19q13.1, which serves as a calcium-release channel for the sarcoplasmic reticulum.5,8,9,10
We describe herein the clinical and pathological features of two Korean families with CCD in whom mutations were identified at the C-terminal region of RYR1. To the best of our knowledge, this is the first genetic analysis of Korean CCD patients.
Case Report
Case 1
A 15-year-old boy presented with slowly progressive, bilateral weakness of the legs that had been present since beginning school. His pre- and perinatal history was unremarkable. Motor milestones were not delayed. He could not run and had experienced difficulty in climbing stairs since childhood. At the time of presentation he had difficulty in arising from a chair. A physical examination revealed diffuse muscle atrophy, especially in the lower extremities. An examination of muscle power disclosed a predominant proximal leg weakness with mild weakness in the bilateral upper extremities and distal legs. The deep tendon reflexes were preserved and there was no facial weakness or skeletal deformities. Serum creatine kinase was within normal limits (106 U/L). Needle electromyography disclosed active myopathic patterns in his limb muscles. Spine roentgenography indicated that scoliosis was not present. The findings of muscle CT and the pulmonary function test were normal.
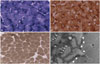
With regard to family history (Fig. 1A), the patient's mother had experienced developmental delay since infancy and sustained mild, nonprogressive proximal leg weakness. She had difficulty in climbing stairs and running. The mother reported that the proband's older sister also had mild, nonprogressive proximal leg weakness and developmental delay. His mother exhibited only mild limb muscle weakness in the proximal legs, without weakness in facial or upper-limb muscles. None of the family members had previously undergone surgery requiring general anesthesia. Muscle biopsy samples taken from the biceps brachii in the proband revealed many peripheral cores with unclear borders in the muscle fibers that were devoid of enzyme activities on nicotinamide dehydrogenase-tetrazolium reductase (NADH-TR) and cytochrome C oxidase (COX) staining. Almost all cores were singular within a muscle fiber, with two cores present in only a few fibers. In addition, all of the muscle fibers were type 1 fibers. Electron microscopy revealed misalignment of myofibrils in the core region compared with those within intact fibers (Fig. 2).
Since screening the entire RYR1 coding region in CCD patients is impractical due to the size of the gene (>159 kb),5 mutation screening was first focused on the mutational hotspots. Since most of the dominant mutations have been identified in the C-terminal region, including exons 101, 102, and 103 of RYR1,1,5,11 these exons were first amplified through PCR. This genetic analysis revealed a novel heterozygous mutation [c.14590T>C (p.Tyr4864His)] in exon 101 (Fig. 1B). This mutation was not detected in 100 control chromosomes by direct sequencing, and the predicted mutant amino acid tyrosine was highly conserved in the RYR1 protein between different species.
Case 2
A 20-year-old man presented with nonprogressive symmetrical upper- and lower-limb weakness since childhood. He had delayed motor developmental milestones and had been unable to walk alone until 24 months of age. Furthermore, his athletic performance at school was worse than those of his peers, and had experienced difficulty in climbing stairs since childhood. A physical examination revealed muscle weakness in the neck and trunk muscles as well as in the proximal upper and lower limbs. His facial muscles were preserved. Gower's sign was positive, and the deep tendon reflexes were symmetrical and normoactive. He had a serum creatine kinase level of 61 U/L. Needle electromyography disclosed positive sharp waves and motor unit potentials with small-amplitude, sharp waves in his upper-limb muscles. Muscle CT revealed atrophy in his hip extensors and bilateral vastus lateralis muscles. No musculoskeletal deformities, such as scoliosis, were observed. The pulmonary function test was unremarkable.
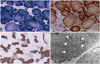
With regard to his family history, his mother had also experienced limb muscle weakness since childhood (Fig. 1A). None of his family members had received general anesthesia. A muscle biopsy sample taken from the vastus lateralis revealed numerous peripheral and central cores, primarily in type 1 muscle fibers on NADH-TR, succinic dehydrogenase, and COX staining. There were more peripheral cores than central cores. Selective type 1 fiber atrophy was also observed without significant fiber-type predominance.
Electron microscopy disclosed disruption of the myofibrillar organization (Fig. 3). Mutational analysis for RYR1 revealed the heterozygous mutation [c.14678G>A (p.Arg4893Gln)] in exon 102 (Fig. 1B). This mutation was previously reported to be associated with the CCD phenotype with AD inheritance.1,12
Discussion
Ryanodine receptor 1 mutations are reportedly responsible for more than 90% of CCD cases.5 The RYR1 mutations associated with CCD or malignant hyperthermia are clustered into four relatively restricted regions, which encode the N- and C-terminal regions, the central portion, and exons 47 and 48.5 Most mutations associated with CCD have been identified in the C-terminal.1,5,11 The C-terminal comprises the transmembrane/luminal and pore-forming regions of the sarcoplasmic reticulum calcium-release channel,6,9 and mutations in this region of the protein may directly alter the functions of the channel, resulting in disruption of calcium homeostasis and/or excitation-contraction coupling.13,14,15,16,17
According to a previous study, there is a link in patients between C-terminal mutations and severe clinical phenotypes, with earlier onset (such as infantile hypotonia) and clearly demarcated central cores in muscle biopsy specimens.5 Patients with mutations outside the C-terminal region are usually associated with milder or asymptomatic clinical phenotypes and peripheral cores in their muscle fibers.5 Wu et al.5 found mutations in the C-terminal domain in 16 of 27 unrelated Japanese patients with CCD. Twelve of these 16 patients presented with symptoms during the perinatal period and had significant joint contracture and scoliosis. Although the mutations identified in the present cases were located at the C-terminal region, all of the affected members exhibited a mild clinical phenotype without facial muscle involvement and skeletal deformities, and atypical, inexplicitly ovoid cores characterized by indistinct borders in the subsarcolemmal areas. As clearly observed in the present cases, a clinicogenetic correlation is not always straightforward in CCD, as was suggested previously.5
To the best of our knowledge, the p.Tyr4864His mutation identified in case 1 of this study is a novel mutation. Another missense mutation affecting the same amino acid (p.Tyr4864Cys) has been reported previously in an English family, who exhibited more severe clinical phenotypes than those in the present family, and significant skeletal deformities such as congenital hip dislocation, lumbar lordosis, thoracic scoliosis, and congenital foot deformity. Three of five members harboring the mutation also had facial muscle weakness.6 A known mutation (p.Arg4893Gln) of case 2 has previously been found in two studies.1,12 A Chinese family with this mutation had no skeletal deformities or respiratory involvement, but had facial muscle atrophy, and their muscle samples exhibited typical central cores.12 Interestingly, a different amino acid change of the same residue (p.Arg4893Trp) presented as respiratory impairment, scoliosis, and multiple mini-cores on biopsy.6
All of our patients had muscle complaints that had been present since childhood. An interesting feature in the family of case 1 is a marked difference in clinical severity between the index case and the other affected family members. Such intrafamilial clinical variabilities have been reported previously in CCD.2,6,11,18 Unfortunately, no muscle or blood samples were available from the parents of the index case for further investigation. The finding that the mother and older sister of the proband also had symptoms suggested that the mutation was initially inherited as an AD trait. However, the severity of the disorder could not exclude the implication of an additional factor, such as a mutation in the paternal allele or a neomutation of the RYR1 gene that has not yet been identified due to the partial screening of the gene in this study. Affected children born from asymptomatic parents have been reported previously who carried a mutation on the maternal allele and another mutation on the paternal allele.2
Muscle biopsies of the index cases revealed numerous peripheral and central cores with deficient oxidative enzyme activities, and electron microscopy disclosed the presence of myofibril misalignment in the presence of mitochondria. Another common feature of CCD-marked type 1 fiber predominance7- was also observed in case 1. According to the literature, the muscle pathology of CCD is highly variable: while some patients exhibit typical central cores, others may instead exhibit peripheral cores or multiple mini cores, and yet others may exhibit no core structures.6,7
We report herein two Korean families with CCD in which a novel missense mutation and a known missense mutation were identified. Although the mutation was located on the C-terminal region, the clinical phenotype was mild, and atypical cores were observed in muscle biopsy specimens. The origin of the phenotypic variability in CCD is still unknown, and further investigations including genetic and functional studies are needed.

 XML Download
XML Download