

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Charcot-Marie-Tooth disease (CMT) is a clinically and genetically heterogeneous disorder of the peripheral nervous system.1 Demyelinating CMT neuropathies are classified as either autosomal dominant CMT type 1 or autosomal recessive CMT type 4 (CMT4).2 CMT4 has been further divided into many subtypes from CMT4A to CMT4J according to clinical symptoms and genetic causes.3 Dejerine-Sottas neuropathy (DSN), which is also known as CMT type 3, is characterized by early age at onset, very slow motor nerve conduction velocities (MNCVs), and severe demyelinating neuropathy.4 Previous reports have described several genes that are relevant to DSN, including those encoding early growth response 2, myelin protein zero, peripheral myelin protein 22, and periaxin (PRX).4,5,6,7
Mutations in PRX are known to cause DSN and CMT4F.6,7 Loss of periaxin function causes instability of Schwann cell myelination, which eventually leads to demyelination and peripheral neuropathy. It has been shown that PRX-knockout mice develop a spectrum of morphologic changes to the myelin sheath.8
Case Report
A 10-year-old girl (Fig. 1A, II-3) was the third child with two healthy older sisters born to nonconsanguineous parents (FC390). Neurologic and electrophysiologic examinations revealed that the parents (I-1 and I-2) and two sisters (II-1 and II-2) did not exhibit distal motor weakness, sensory deficit, or abnormal electrophysiologic findings. However, the patient had delayed motor milestones, with autonomous walking not present until 24 months of age. During childhood she developed gait difficulties with clumsiness and balance problems; however, the disease progression was very slow. A neurologic examination at 10 years of age revealed bilateral pes cavus, scoliosis, and a broad-based gait. She had severe weakness of foot eversion without proximal muscle involvement. Her sensitivities to pinprick, touch, position, and vibration were decreased; the vibration sensation was more severely disturbed than pain sensations in all limbs. Sensory ataxia and a positive Romberg sign were present. Deep tendon reflexes were absent and pathologic reflexes were not found. Her CMT neuropathy score was 20.15
Electrophysiologic studies were performed at 7 and 10 years of age. Sensory nerve action potentials were absent in the median, ulnar, and sural nerves. Compound muscle action potentials were severely reduced in the median nerve (range, 0.2-0.5 mV) and absent in the ulnar, peroneal, and tibial nerves. Median MNCVs were markedly reduced (range, 2.6-3.0 m/s), and distal latencies were dramatically prolonged (range, 39.6-44.8 ms). The needle electromyogram was consistent with chronic neuropathy.
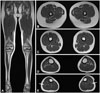
MRI scans of the hip and thigh were normal (Fig. 2A, B, and C). Lower-calf MRI revealed hyperintense signal abnormalities (Fig. 2D and E). T1-weighted images demonstrated mild signal changes and atrophy in the peronei muscles; however, the tibialis anterior and soleus muscles were not involved.
The results of a histopathologic study performed at age 10 years are summarized in Table 1. Light-microscopy examination of longitudinal and cross-sections of nerve fibers revealed nerve fascicles that were markedly decreased in size, diffuse subendoneurial edema, and moderately to markedly decreased numbers of myelinated fibers (MFs) of all calibers, with suggestion of onion-bulb formation and endoneurial fibrosis (Fig. 1C). The mean diameter of MFs (3.14 µm) and the percentage area of MFs (1.39%) were also lower than in age-matched controls (4.5 µm and 25.2%, respectively) (Fig. 1D). Electron-microscopy examination revealed findings consistent with abnormal myelin, such as focally folded, uncompacted, or deteriorating myelin, irregular myelin thickness, or fragmented myelin structures in Schwann cells or nearby macrophages (Fig. 1E and F).
Mutational analysis
Exomes were captured using a SeqCap EZ device (version 3.0, Roche-NimbleGen, Madison, WI, USA), and sequencing was performed using a genome analyzer (HiSeq 2000, Illumina, San Diego, CA, USA). The University of California, Santa Cruz assembly hg19 (National Center for Biotechnology Information build 37.1) was used as the reference sequence. The candidate variants considered to be causative were confirmed by Sanger sequencing. The total sequencing yield of WES was approximately 10.47 Gbp, with a 91.72% coverage rate for the targeted exon regions (≥10×). The total number of observed variants was 69,592 single-nucleotide polymorphisms, of which 349 variants were observed in CMT-relevant genes that have been reported previously.16 Subsequent filtering was achieved for functionally significant variants, where the functionally significant variants indicate all variants that change the amino acid, such as nonsynonymous, splicing site, stop-gain/-loss, and coding indels. Filtering isolated 25 functionally significant variants in the CMT-relevant genes. Most variants have been reported in the dbSNP137 or 1000 Genome databases, except for three variants [one in the gene encoding dynactin subunit 1 (DCTN1) and one in PRX]. Mutation in DCTN1 c.2054T>G (p.V685G) was excluded because the variant was detected in unaffected family members (father and an elder sister) as well as a healthy control by capillary sequencing. However, a pair of compound heterozygote nonsense mutations, c.1174C>T (p.R392X) and c.2035C>T (p.R679X), in exon 7 of PRX were well fitted with an autosomal-recessive inheritance model within the family, and neither was found in normal controls (n=300). The c.1174C>T and c.2035C>T mutations were inherited from the patient's mother and father, respectively (Fig. 1B). Thus, it was postulated that both mutations lead to enzymatic defects, and that these compound heterozygous mutations were the underlying cause of the DSN phenotype in this patient.
Discussion
We have identified novel compound heterozygous mutations in PRX from a Korean DSN family. The phenotype of the affected patient was similar to that of previously described PRX cases,10,11,12,13,14 with early age at onset, sensory ataxia, very slow MNCVs, and severe demyelinating neuropathy consistent with DSN. One of the R392X and R679X mutations was detected in each of the patient's asymptomatic parents, and neither of the mutations was observed in the 300 healthy controls.
Most reported causative PRX mutations have been nonsense or frameshift mutations, and the novel mutations reported here in are also nonsense mutations.5,6,7,10,11,12,13,14 One exception was reported recently in a Japanese patient with a missense mutation in PRX;9 that patient exhibited an adult-onset phenotype that was milder than in patients with nonsense mutations.
It is well known that PRX-associated neuropathies are characterized by slow clinical progression.7 In spite of markedly severe electrophysiologic defects, it is surprising that the present patient was able to walk without orthopedic foot devices at 10 years of age and had exhibited very slow disease progression. Lower-limb MRIs revealed only mild fatty infiltrations of the bilateral peronei muscles, but the other muscles of the lower extremities were not involved. These MRI findings represented evidence of a mild phenotype, which was consistent with the clinical manifestations.
To the best of our knowledge this is the first patient of Korean origin with a PRX mutation. PRX mutation screening is recommended in Korean patients with early onset and slowly progressive demyelinating neuropathies.


 XML Download
XML Download