

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Most cases of amyotrophic lateral sclerosis (ALS) are sporadic, and approximately 5% of ALS patients are known to have a familial form (fALS). Among fALS patients, mutations in the gene encoding Cu/Zn superoxide dismutase (SOD1) are the most common cause, accounting for approximately 20% of fALS cases in Korea.1
While the underlying causes of ALS and the pathogenic mechanisms underlying the SOD1 mutation have yet to be fully elucidated, there is accumulating evidence that immune-system alterations play an important role in noncell autonomous toxicity in ALS with SOD1 mutations in both human and animal models. The unusual overlap of ALS with numerous autoimmune diseases supports an immune-related pathological mechanism in ALS.2
Bullous pemphigoid (BP) is an autoimmune inflammatory disease characterized by tense, subepidermal bullae that develop on normal or erythematous skin, as well as linear immunoglobulin G (IgG) autoantibodies and C3 deposition at the dermal-epidermal junction.3 It has been suggested that neurodegenerative processes may trigger the development of BP through an autoimmune mechanism in patients with various neurological diseases.4,5 Among the co-occurrences, a few cases of BP and ALS coexistence have been reported,3 but the association between BP and ALS has not been clearly explained.
Herein we report a patient with a novel codon mutation [phenylalanine 45 to serine (F45S)] in SOD1 who developed BP over the entire body. We hypothesize that the SOD1 mutation of ALS is associated with the development of BP.
CASE REPORT
A 57-year-old male with a family history of ALS (in his mother and younger sister) presented with progressive muscular weakness (Fig. 1A). He first noticed weakness in his right upper limb at the age of 55 years. Eighteen months later we performed electrophysiological studies on the patient, who exhibited symptoms compatible with ALS: active denervation and reinnervation potentials in three spinal regions (cervical, thoracic, and lumbosacral) and the right trapezius using needle EMG. Motor-nerve conduction studies revealed decreased compound muscle action potential amplitudes with normal sensory conduction velocities. There was a mild weakness of both the upper limbs [Medical Research Council (MRC) grade=4/5] and the lower limbs (MRC=4/5), with mild atrophic changes. The patient was hyperreflexic in both the upper and lower limbs; Hoffmann signs were present on the right hand, but a bilateral Babinski sign was absent. Sensory functions were normal. Biochemical and immunological screening ruled out ALS-mimic syndrome. MRI findings of the brain and cervical spine were normal. No skin lesions were present on the patient, his mother, or his younger sister.
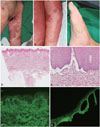
Twenty-two months later the patient had a severe functional disability of gastrostomy and placement on a tracheostomy-assisted ventilator, and he suddenly presented with erythematous patches studded with small vesicles on both his hands and soles. After 1 week, large, tense bullae on apparent erythematous patches developed on the hands, feet, and axillae (Fig. 2A-C). A histopathological examination revealed subepidermal blistering with an inflammatory infiltrate composed of numerous eosinophils in the upper dermis. Direct immunofluorescence testing revealed a linear pattern of IgG and C3 deposition at the dermal-epidermal junction. Indirect immunofluorescence testing with 1-M-NaCl-split skin revealed a linear IgG deposit along the epidermal roof of the basement membrane zone up to a titer of 1:160 (Fig. 2D-G). These findings confirmed the diagnosis of BP.
Treatment for BP began with 60 mg/day prednisolone and 1 g/day mycophenolate mofetil, which resulted in the disappearance of the bullous lesions and reduced development of new lesions; then, the prednisolone dose was slowly tapered. The family history based on interviews with the patient and his relatives was suggestive of an autosomal-dominant inheritance. Direct sequencing analyses of SOD1 in the patient using a previously described method1 revealed a novel missense mutation (c.137T>C; F45S). The patient's mother had died at age 72 years, only 1 year after being diagnosed with ALS, while his younger sister had died 8 months after being diagnosed with ALS, at the age of 52 years (Table 1). In a follow-up study we investigated his 48-year-old youngest sister and 31-year-old daughter after consent for genetic examinations had been obtained; both individuals carried this same SOD1 mutation(F45S)(Fig. 1B). Both individuals remain unaffected.
DISCUSSION
We have identified a fALS patient with a novel missense mutation at codon 45 of SOD1, wherein phenylalanine was converted to serine (F45S) instead of cysteine (F45C). In sharp contrast to the phenotype of fALS with SOD1 (F45C), which shows a slow progression with mild functional impairment,6 the phenotype of fALS with the F45S mutation in the present case exhibited rapid progression of weakness, resulting in a completely dependent functional status within 1-2 years. The observed variability in the clinical phenotype dependent on the substituted amino acid at the same codon of SOD1 was consistent with previous findings.1,7
Interestingly, we found BP in the skin of the fALS patient; this is an autoimmune blistering skin disease in which autoantibodies develop against the structural proteins of the dermal-epidermal junction [BP antigen 1 and 2 (BPAG 1 and 2, respectively)]. To the best of our knowledge, BP in an fALS patient with the SOD1 mutation has not previously been reported, despite there being two reports, without genetic information, on the occurrence of BP in ALS patients.3
Previous studies have found BP to be associated with a chronic bedridden state, medication, and genetic and environmental factors.5,8,9 The patient in the present case was nearly quadriplegic (MRC=0 or 1/5) and in a completely bedridden state. Although there was no association between the regional involvement of BP and the preferentially affected region, the patient's severe disability throughout the body and limbs were associated with skin lesions on abdomen, chest, and along the entire limb. At the time of BP development the patient had been taking the following medications for at least 2 months: acebrophylline, acetylcysteine, vitamin B complex, ranitidine, ubidecarenone, tocopherol, and tamsulosin. Although there are no reports of associations between BP and these drugs,4,5 we could not rule out the potential adverse effects of these particular individual drugs or combinations thereof.
We hypothesized that SOD1 with the F45S mutation plays a role in the overlap between ALS and BP through common mechanisms based on the following facts. First, the pathogenesis of both BP and ALS with the SOD1 mutation could be related to the alteration in the immune reaction. BP is an autoimmune disease, as suggested by the presence of the autoantibodies BPAG 1 and 2, and the dysfunction of regulatory T cells (Treg cells); it is associated with a wide range of autoimmune diseases in the neuromuscular system, such as multiple sclerosis and myasthenia gravis. ALS patients with the SOD1 mutation exhibit hallmarks of autoimmunity such as the presence of circulating immune complexes and dysfunction of the Treg cells.2 These features raise the possibility of an overlap between BP and ALS in terms of autoimmunity. Second, matrix metalloproteinase-9 (MMP-9) is a common factor linking BP and ALS with the SOD1 mutation. MMP-9 has been suggested to play a necessary role in the formation of subepidermal blistering, as evidenced by findings of MMP-9-deficient mice being resistant to blister induction. ALS patients with the SOD1 mutation exhibited an increased level of MMP-9 in the central nervous system and the skin.10 Finally, the changes in cytoskeletal structure in SOD1 transgenic mouse models suggest the occurrence of BP. Impairment of cytoskeletal structures including neurofilaments in the SOD1 transgenic mouse model3 and BP support this hypothesis.3
In conclusion, this is the first report of an ALS patient with a novel codon mutation, F45S, in SOD1. The patient simultaneously presented with BP coexistence. In addition to its role as a potential predisposing factor for BP, particularly for the development of BP in SOD1-mutated ALS patients, we suggest that the SOD1 mutation altered the immune response, including dysregulation, changed levels of MMP-9 or the cytoskeletal structure. This suggests that diseases featuring an autoimmune response tend to be associated more often than can be ascribed to chance; therefore, further molecular approaches regarding the genetic-immunological linkage between the two traits are required.


 XML Download
XML Download