

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Duchenne and Becker muscular dystrophies (DMD and BMD, respectively) are the most common X-linked recessive muscular dystrophies caused by mutations in the dystrophin gene that encodes a muscle cytoskeletal protein.1 Muscular dystrophy is also called dystrophinopathy because it is caused by mutations in the DMD gene-which encodes dystrophin-at the Xp21 locus.2 DMD is the more severe form of dystrophinopathy and is caused by a frameshift mutation.1 Patients with BMD usually carry in-frame mutations that lead to amino acid translations, and therefore manifest a milder phenotype that is characterized by muscle weakness, exercise intolerance, myoglobinuria, and myalgia.1 DMD and BMD usually affect men, but women also occasionally develop symptoms, and two-thirds of mothers of affected males are thought to be DMD carriers.3
Reportedly 2.5-7.8% of female DMD carriers report muscle weakness and are therefore classified as manifesting DMD carriers.234 Hoogerwaard et al. found that 5% of female DMD carriers present with regular myalgia and cramps without muscle weakness, 17% experience mild-to-moderate muscle weakness, and 8% present with dilated cardiomyopathy, with an average onset age of 33 years.5 Since most heterozygous female carriers of DMD have subclinical symptoms, screening of dystrophinopathy carriers may only be performed on clinical grounds such as a clear X-linked family history of muscular dystrophy.6 However, myopathic muscle biopsy and advanced molecular diagnostic analysis can be used to identify DMD carriers who do not have a positive family history of dystrophinopathy; 10% of women with abnormally elevated serum creatine kinase (CK) are DMD carriers.3 The clinical characteristics of genetically confirmed female dystrophinopathy carriers were explored in this study.
METHODS
The database records from Gangnam Severance Hospital in South Korea collected between July 2007 and October 2013 were searched for women with clinical features indicative of dystrophin gene mutations. As in previous studies,5789 female carriers were classified as symptomatic when they manifested symptoms of muscle weakness and/or dilated cardiomyopathy, and as asymptomatic when they exhibited high serum levels of CK and/or minor myopathic signs such as muscle cramps and myalgia, but did not have muscle weakness or dilated cardiomyopathy.5 Isolated CK elevation alone would not be sufficient to consider a patient as a manifesting carrier.9 A diagnosis of dystrophinopathy is usually considered after careful review of the subject's clinical features, family history, and laboratory evidence of elevated serum CK. Cases were confirmed through examinations such as muscle biopsy or molecular genetic testing. DMD mutation analysis employing a combination of techniques was applied to DNA extracted from peripheral blood.8 Intragenic deletions and duplications were analyzed using [multiplex ligation-dependent probe amplification (MLPA); MRC-Holland, Amsterdam, The Netherlands].10 The following data were collected retrospectively for symptomatic female carriers: clinical features, ages at onset and diagnosis, serum CK levels, and findings of cardiology tests, electromyography, muscle MRI, and muscle biopsy analysis.
RESULTS
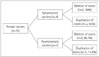
Of the 285 patients (230 men and 55 women) who were screened for dystrophin gene mutations, 104 men and 12 women with mutations were identified; 1 symptomatic female carrier who also had Turner syndrome was excluded. Among the included 11 female carriers, 4 were symptomatic and 7 were asymptomatic (Fig. 1). The most common reason for asymptomatic female carriers visiting the hospital was a family history of muscular dystrophy. The clinical presentations and detected gene mutations in symptomatic female carriers are summarized in Table 1.
Two of the symptomatic women had out-of-frame deletions and two had duplications. The mutation sites in both patients with deletions were distributed in hot-spot lesions (exons 2-20 and 44-53). Of the seven asymptomatic female carriers, six had out-of-frame deletions and only one had a duplication (Fig. 1). In the symptomatic dystrophinopathy carriers, the age at symptom onset varied from 15 to 31 years (mean, 30.6 years), and the age at diagnosis varied from 30 to 35 years (mean, 34.5 years). The ages at diagnosis in the seven asymptomatic carriers were 4, 22, 26, 27, 27, 28, and 38 years (mean, 24.5 years). Serum CK levels were elevated (mean, 1,301 IU/mL normal range, 35-232 IU/mL) in three of the four (75%) symptomatic female carriers, but only mild increases (mean, 347 IU/mL) were noted in three of the seven (42%) asymptomatic female carriers.
DISCUSSION
Several mechanisms have been proposed to explain symptom manifestation in women with DMD/BMD.56789 The most frequently reported mechanisms are skewed X-chromosome inactivation (XCI), in which expression of the X chromosome with the DMD mutated allele is favored,11 and balanced X-autosome translocation.12 The relationship between XCI and clinical severity is not clear, and the prognostic value of XCI is controversial.11 As observed in the present study, some women with Turner syndrome have a dystrophin mutation on the remaining X chromosome,13 and others have a dystrophin mutation on each X chromosome. There are also rare cases of dystrophin mutations in females with X chromosome uniparental disonomy,14 and in male pseudohermaphrodites with mutations in the androgen receptor gene.15
The diagnostic approach to female dystrophinopathy includes a clinical history of myopathic symptoms and signs as well as a family history. The presence of elevated serum CK is an important indicator of carrier status, but this occurs in only about 70% of carriers, so a normal CK level does not exclude the possibility of being a carrier. Electromyography, MRI analysis, muscle biopsy with dystrophin immunostaining, and DNA analysis are the predominant tests used for determining the presence of dystrophinopathy. The exclusion of other types of neuromuscular diseases is also important for diagnosing dystrophinopathy.
Dystrophin gene mutation analysis reveals a deletion of one or more exons in 60-70% of dystrophinopathy cases.1016 DMD gene duplications account for 5-15% of DMD cases, and point mutations or small deletions/insertions account for 25-30%.1617 The multiplex polymerase chain reaction is a common technique used to identify DMD gene mutations, and it can detect approximately 98% of deletions; however, it is not useful for detecting duplications or identifying female carriers.10 Conversely, MLPA can detect the deletions and duplications in both male and female carriers, and so this procedure is currently the gold standard for DMD gene molecular analysis.10 Unlike male DMD patients, manifesting female carriers have variable disease activity and can even be asymptomatic, and so their status can go undetected.18
The symptomatic female carriers in this study were characterized clinically and genetically. First, the age at symptom onset in these patients was addressed. In previous studies the mean symptom onset age for female carriers was 33.6 years,5 compared to 39.6 years for cardiomyopathy.19 In contrast, male patients with DMD exhibit their first symptoms at 3.0±1.8 years (mean±SD), while patients with DMD varied in their symptom presentation (mainly elevated serum CK, weakness, fatigue, myalgia, or cramps) at the age of 12.9±11.8 years.2021 In the present study, the age at symptom onset among the female carriers ranged from 15 to 31 years (mean, 30.6 years), and the age at diagnosis for asymptomatic carriers ranged from 4 to 38 years (mean, 24.5 years). Muscular dystrophy is a progressive disease, and the age at symptom onset in female carriers is far older than would be expected for homozygous male patients carrying the same mutation. The present findings suggest that asymptomatic female carriers should be followed up because they were younger at diagnosis than the age at symptom onset in symptomatic carriers, and most were evaluated before pregnancy.
Elevated serum CK levels were found in the present symptomatic female carriers, while only asymptomatic female carriers exhibited mildly elevated CK levels. Serum CK measurement is the most commonly used method for carrier detection,22 and elevated levels are found in up to 50% of carriers.5 Interestingly, all symptomatic female carriers in the present study manifested bilateral leg weakness as the initial symptom. Consistent with previous reports,723 three of the four patients (75%) reported asymmetric muscle weakness, which has previously been reported as being present in between 15% and 81% of symptomatic carriers.3523 Muscle MRI in female carriers is more sensitive than clinical examination for detecting single-muscle involvement and asymmetry.7 Finally, symptom severity-exacerbated muscle weakness-in the symptomatic female carriers was aggravated after giving birth (Table 1). Given that dystrophinopathy is typically studied in males, the association between female dystrophinopathy and labor has not previously been explored, and asymptomatic female carriers should be closely monitored for symptoms during and after labor.
The findings of this study should be interpreted with caution in the light of certain limitations. For example, relatively few patients were included and the retrospective design of the study meant that chromosome or XCI studies could not be performed. Further studies with larger samples are needed to determine whether the present findings are generalizable to broad patient populations.
Female symptomatic dystrophinopathy is a rare condition whose diagnosis can be challenging. The MLPA method is a simple, rapid, and reliable tool for screening for DMD/BMD gene mutations. Since female dystrophinopathies are important factors that affect the prevalence of DMD/BMD in males, early diagnosis of DMD/BMD and appropriate genetic counseling are needed. In conclusion, understanding and characterizing female dystrophinopathy is helpful for the establishment of diagnostic approaches in patients with a negative family history who may have been wrongly diagnosed with limb-girdle muscular dystrophy, inflammatory myositis, or an unknown myopathy.

 XML Download
XML Download