

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Horner syndrome (HS), also known as Claude-Bernard-Horner syndrome or oculosympathetic palsy, comprises ipsilateral ptosis, miosis, and facial anhidrosis. This article describes a case of congenital HS with heterochromia iridis associated with ipsilateral hypoplasia of the internal carotid artery (ICA), confirmed by computed tomography (CT) analysis of the skull base.
CASE REPORT
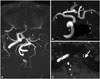
A 67-year-old man underwent magnetic resonance imaging (MRI) for occasional headaches. He had high blood pressure and hypercholesterolemia, was obese, and had previously smoked cigarettes, and had been treated with perindopril, amlodipine, low-dose acetylsalicylic acid, and simvastatin. MRI revealed chronic ischemic injuries in the left superficial middle cerebral artery (MCA) territory, associated with an extremely spindly left ICA, which was misinterpreted as ICA thrombosis (Fig. 1). The left anterior cerebral artery and MCA were supplied by a large posterior communicating artery from the basilar artery.
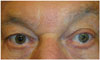
This patient was examined in the neuro-ophthalmology department of our hospital, where heterochromia iridis and miosis were reported in the left eye in comparison with the right: the left pupil was 1.5 times smaller than the right upon exposure to light. The ptosis of the left upper eyelid was barely discernible because of frontal compensation (Fig. 2): the left and right palpebral fissure heights were symmetric, with bilaterally symmetric inferior scleral show (no inverse ptosis), but the distance between the left upper eyelid and the left eyebrow was 1.5 times larger than that on the opposite side due to elevation of the left eyebrow. The left pupil did not dilate after administering 5% cocaine eyedrops, confirming the presence of HS. The patient reported that these anomalies had always been present, and a diagnosis of congenital HS was reached. Unenhanced multidetector CT of the skull base demonstrated a left carotid canal hypoplasia that was consistent with congenital hypoplasia of the left ICA (Fig. 3). This congenital anomaly did not require any specific treatment other than strict control of cardiovascular risk factors. The patient denied a family history of any similar disorder.
DISCUSSION
Changes in pupil size are controlled by the parasympathetic (pupilloconstrictor) and sympathetic (pupillodilatator) nervous systems.1 HS is provoked by a deficiency of at least one of the three types of neuron in the ipsilateral oculosympathetic pathway, and can be acquired, congenital, or (rarely) hereditary.1,2 Injury of the sympathetic system may have a central origin, by involving the hypothalamospinal tract (e.g., trauma of the cervical spinal cord, medullary tumor, syringomyelia, neuroblastoma, or congenital cytomegalovirus infection), and may affect the preganglionic neurons of the sympathetic chain [e.g., lung apex tumor (Pancoast syndrome), goiter or thyroid carcinoma, or iatrogenic sympathectomy or birth trauma with traction on the neck during delivery] or the postganglionic neurons at the level of the ICA (e.g., carotid artery dissection, carotid artery agenesis, or cervical lymph node compression).
Agenesis, aplasia, and hypoplasia of the ICA are very rare congenital anomalies, occurring in less than 0.01% of the population.3 Agenesis is defined as a complete failure of ICA development, unlike aplasia and hypoplasia, which are respectively total or partial nondevelopment of the ICA despite the presence of an existing embryonic precursor of the vessel.4 The precise mechanisms underlying these developmental anomalies are unknown. Associations with corpus callosum agenesis, meningocele, neurofibromatosis, Klippel-Feil syndrome, 22q11.2 deletion syndrome, coarctation of the aorta, and other cardiac anomalies have been described.5
Congenital absence of the ICA (through agenesis, aplasia, or hypoplasia) may be unilateral (>90%, with a nearly 3:1 left-sided predominance) or bilateral,3,6 and can be complete or may involve only a segment of the artery. Most patients are asymptomatic due to sufficient development of the collateral blood flow. The most frequent type of collateral flow is through a functional Willis polygon; less frequently the collateral flow is provided by transcranial collaterals from the external carotid artery or via persistent embryonic vessels.3,6 However, these patients may present later in life with symptoms related to cerebrovascular insufficiency, with the risk increasing with age.6 Alternatively, these patients have an increased risk of developing cerebral aneurysms (reported prevalence of 24-34% versus 2-4% in the general population) and subarachnoid hemorrhage due to increased blood flow through collateral vessels and altered flow dynamics.3,6,7
The first case of ICA agenesis associated with congenital HS was reported in 2000 by Ryan et al.,8 since when several similar associations have been described. Many cases of ICA agenesis have been reported since 1954 without mention of associated HS, leading Ryan et al.8 to hypothesize that sympathetic fibers can establish alternate routes during development. However, to the best of our knowledge, ICA agenesis with confirmed absence of HS has not yet been described. We therefore disagree with Ryan et al., and believe that ICA agenesis is probably always associated with congenital HS, due to the impossibility of development of orthosympathetic fibers around the missing ICA. It is possible that authors who previously reported ICA agenesis without evocation of associated HS did not actually look for HS (articles published before 2,000 were based on multiples of retrospective angiographic analyses or postmortem examinations, most often without clinical correlations); after all, we only see what we look for.
Our review of the literature and our personal experience revealed two main clinical presentations of HS due to ICA anomalies. The first such cases most often occur in young patients, in which the evaluation of asymmetric pupil size or heterochromia iridis has led to the diagnosis of congenital HS, and the subsequent examinations have revealed secondarily an associated ICA agenesis or hypoplasia. The second case type is most common in older patients, with symptoms due to cerebrovascular insufficiency or subarachnoid hemorrhage leading to the diagnosis of ICA anomaly, and watchful physical examination secondarily revealing the congenital HS with heterochromia iridis. In this latter presentation, congenital HS may be unrecognized due to the predominance of other neurological symptoms.
CT evaluation of the skull base is essential: ICA agenesis is confirmed by the absence of the carotid canal, whereas aplasia or hypoplasia of the ICA results in a carotid canal hypoplasia.3,4,5,6 Indeed, the embryological development of the carotid canal requires the presence of the ICA or its precursor.5 Moreover, differentiation between the absence or narrowing of the carotid canal allows us to distinguish congenital absence of ICA from acquired causes of ICA obstruction or stenosis (e.g., thromboembolism, severe atherosclerosis, arterial dissection, or fibromuscular dysplasia). These distinctions are compulsory due to their different therapeutic implications.
Heterochromia iridis refers to a difference in coloration of the iris due to asymmetric concentration or distribution of melanin in the iris tissues, and can be complete or partial (sectoral), and congenital (inherited as an autosomal-dominant trait or due to genetic mosaicism) or acquired.9,10 Congenital heterochromia can be caused by iris hamartomas (in neurofibromatosis), ocular melanosis, oculodermal melanocytosis (nevus of Ota), pigment dispersion syndrome, or Sturge-Weber syndrome (with abnormal hyperpigmentation of the iris), or by isolated iris hypoplasia, congenital HS, Waardenburg syndrome, piebaldism, Hirschsprung disease, incontinentia pigmenti, or Parry-Romberg syndrome (progressive hemifacial atrophy)-with an abnormal hypopigmentation of the iris. Acquired heterochromia can be provoked by injuries, inflammation (e.g., chronic iritis or Fuchs heterochromic iridocyclitis), iron deposition (siderosis or hemosiderosis), tumors (e.g., melanomas, metastasis, leukemia/lymphoma, or juvenile xanthogranuloma), the use of certain eyedrops (e.g., prostaglandin analogues), or (rarely) by different syndromes (e.g., iridocorneal endothelium syndrome, iris ectropion syndrome, acquired HS, or Duane syndrome).9,10
In HS, the deficiency of sympathetic activity can interfere with melanin pigmentation of the melanocytes in the superficial stroma of the iris. In this context heterochromia iridis is observed in congenital HS, and also in HS that occurs in children younger than 2 years of age and, exceptionally, in long-standing HS.1

 XML Download
XML Download