

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Muscular dystrophy is a clinically and genetically heterogeneous inherited disorder characterized by progressive muscle weakness and wasting. A step-by-step approach with assessment of medical history, clinical examination, laboratory evaluation, muscle pathology, muscle immunoanalysis, and mutational analysis is typically used for the diagnosis of muscular dystrophy.1,2 However, this serial approach often fails to identify causative mutations due to high phenotypic and pathologic variability, small pedigrees, and the limited power of traditional linkage analyses.3 Recent advances in next-generation sequencing has made it possible to selectively sequence only the protein-coding exons of the genome, a process termed 'whole exome sequencing' (WES). The application of WES not only saves time but is also cost-effective for the identification of causative genes in Mendelian diseases.4 Therefore, WES is being increasingly adopted for the identification of causative genes in muscular dystrophy research.5,6,7,8
Herein we report a mutation in the gene encoding collagen type VI α1 (COL6A1) in a large Korean family with autosomal-dominant Bethlem myopathy that was detected using WES.
CASE REPORT
Eighteen members of a large Korean family with dominantly inherited myopathy (7 affected and 11 unaffected) were enrolled (Fig. 1A). Written informed consent to participate was obtained from all participants and from the parents of participants younger than 18 years, according to a protocol approved by the Institutional Review Board for Ewha Womans University Mokdong Hospital, Seoul, Korea.
Patients
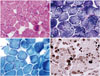
The proband, a 38-year-old woman (III-4), presented with progressive proximal weakness and ankle contractures (Supplementary Table 1 in the online-only Data Supplementary). Her initial development after birth was reportedly normal, but she did not begin walking until she was 16 months old. She recalled that she had always been weaker than her peers. Her motor function remained relatively stable until her mid-20s. However, she experienced slowly progressive muscle weakness after the delivery of her first child at an age of 27 years. Her neck flexors [Medical Research Council (MRC) grade 3] appeared to be more damaged than her neck extensor (MRC grade 4+). The proximal muscles of her upper and lower limbs were more severely involved than the distal muscles, and her ankle joints were affected by contracture. In addition, she appeared to exhibit mild facial weakness and absent tendon reflexes in the upper and lower limbs. However, sensory examination revealed no abnormalities. Laboratory studies revealed a serum creatine kinase level of 66 IU/L (normal, <135 IU/L), and her vital capacity was 3,120 mL. Electrocardiography and echocardiography findings were normal. Nerve conduction studies and needle electromyography revealed active generalized myopathy. A muscle biopsy sample obtained from the left biceps brachii revealed nonspecific muscular dystrophic changes. Hematoxylin and eosin staining revealed variation in muscle fiber size (Fig. 2A), and modified Gomori trichrome staining revealed a few ragged red fibers (Fig. 2B). Architectural changes of disorganized intermyofibrillar networks, such as lobulated fibers, were accentuated in staining with nicotinamide adenine dinucleotide tetrazolium reductase (Fig. 2C). In addition, adenosine triphosphatase (pH 9.4) staining demonstrated a 69% predominance of type I fibers (Fig. 2D). Electron microscopy revealed many mitochondria, and immunohistochemical analyses of the muscle specimens revealed normal staining patterns for the C-terminal of dystrophin, rod domain of dystrophin, N-terminal of dystrophin, dysferlin, α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan, δ-sarcoglycan, α-dystroglycan, and caveolin.
The other affected members of the family had similar clinical presentations (Supplementary Table 1 in the online-only Data Supplementary). They experienced very slow progressive muscle weakness and lived without significant disability until old age. Subject II-2 required aid for ambulation after the age of 50 years, while subject II-3 (61 years old) was able to walk independently. The ankle joints were affected by contracture in all seven affected patients, while the elbow joints were involved only in subject II-2. Based on the clinical and pathologic features, the family was initially diagnosed with autosomal-dominant limb-girdle muscular dystrophy.
After identification of the causative gene, all affected members underwent a second neurologic examination. This identified contracture of the interphalangeal joint-which is a characteristic sign of Bethlem myopathy-in five family members (II-2, II-3, III-4, III-6, and IV-7). These contractures were very subtle and were not found on routine neurologic examination; they were only apparent when the wrist and fingers were extended passively.
Genetic analysis
Whole exome sequencing was performed for five members of the family, including four affected members (II-3, III-4, IV-7, and IV-8) and one unaffected member (IV-6) to identify the genetic causes of the disease, following the method described by Choi et al.9 The exome sequencing data are summarized in Supplementary Table 2 (in the online-only Data Supplementary). The mean total sequencing yield was 9.3 Gbp/sample, and the coverage rate of the targeted exon regions (≥10×) was 93.56%. The average read depth of the target regions was 69.3 reads, and the average number of observed variants per sample was 92,174 SNPs and 9,321 indels. By comparing the exome data between 4 affected and 1 unaffected family members, we found that 15 functionally significant cosegregated variants (Supplementary Table 3 in the online-only Data Supplementary). Subsequent capillary sequencing analysis of control samples and other family members who were not included in the exome sequencing excluded most variants as the underlying cause of myopathy. However, a c.1056+1G>A splicing-site mutation in COL6A1 completely cosegregated with affected status within the family (Fig. 1B), and was not found in 200 healthy controls. This mutation has been reported to be the underlying cause of Bethlem myopathy.10,11,12,13 Thus, we determined that the c.1056+ 1G>A mutation in COL6A1 was the underlying cause of the disease in this family.
DISCUSSION
Whole exome sequencing of five members from a single family identified a splice donor site mutation at c.1056+1G>A of COL6A1. This mutation causes the formation of abnormal collagen VI protein by skipping of exon 14 and consequent in-frame deletion of amino acids from the triple helical domain of the α1 chain.12
Bethlem myopathy is a dominantly inherited myopathy caused by mutations in one of three genes encoding collagen type VI alpha (COL6A1, COL6A2, and COL6A3).14 The phenotype is characterized by slowly progressive proximal weakness and multiple contractures. Prominent contracture in the early stages of the disease is one of the most important clinical features in Bethlem myopathy, Emery-Dreifuss muscular dystrophy, and Ullrich congenital muscular dystrophy. Among these conditions, Bethlem myopathy demonstrates the most benign clinical course and mildest contractures.
Bethlem myopathy is often difficult to diagnose and its frequency may be underestimated for several reasons. First, mild contractures often lead to confusion in the diagnosis.15,16 In the present family, even though ankle contracture was initially detected, this is a common nonspecific finding in many other neuromuscular diseases. Contracture of the interphalangeal joint is a hallmark of Bethlem myopathy, but is often so subtle that it goes unrecognized. Second, muscle biopsy is not typically used for confirmatory diagnosis of Bethlem myopathy due to nonspecific myopathic changes and lack of detected abnormalities of collagen VI, even in immunohistochemical analyses. Both Ullrich congenital muscular dystrophy and Bethlem myopathy are collagen-IV-related myopathies. Immunohistochemistry in Ullrich congenital muscular dystrophy exhibits complete absence or unequivocal reduction of collagen VI compared to normal; it can thus be used for diagnostic purposes. However, in Bethlem myopathy, immunostaining of muscle biopsy with various collagen VI antibodies is usually normal.14 Third, identification of the causative genes by general sequencing is costly and time-consuming because it is necessary to screen all 107 exons in all 3 genes for molecular genetic diagnosis. For these reasons, careful clinical assessment and cost-effective, time-saving strategies for genetic analysis are important for the diagnosis of Bethlem myopathy.
Whole exome sequencing is a well-justified strategy for discovering the causative genes of muscular dystrophy. WES is based on next-generation sequencing, which reduces the cost and time relative to Sanger sequencing.17 In addition, WES focuses only on protein-coding regions, but it is still an effective diagnostic tool because more than 90% of the pathogenic mutations for Mendelian disorders are found in exons.4
In conclusion, we identified a COL6A1 mutation in a Korean family with Bethlem myopathy; this is the first such report in Korea. Even though the causative mutation identified in the present study has been reported previously, this work underscores the usefulness of WES for the diagnosis of muscular dystrophy.

 XML Download
XML Download