

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
A stroke is a major cerebrovascular event that can cause serious disability and even death. The protein expressions induced by stroke in affected brain regions during both the ictal and postictal stages remain a matter of controversy. The concept of proteomes was first introduced in 1995.1 Since then, two-dimensional gel electrophoresis (2-DE) and mass spectrometry (MS) have been used widely to analyze thousands of proteins in individual experiments and to compare the overall protein expressions of cells under different conditions.2,3 Several recent studies have investigated the alterations in protein expression relevant to various diseases, such as certain psychiatric disorders,4 Down's syndrome,5 Huntington's disease,6 and Alzheimer's disease.7 However, one study investigated the proteome of brain microdialysate obtained from the nonlesioned contralateral sides of the brains of three stroke patients.8
Focal brain infarcts are surrounded by extended perilesional zones that comprise the partially ischemic penumbra and nonischemic cortex referred to as the remote area. Little is known about protein expression in the penumbra and remote zones. Dynamic spatiotemporal patterns of gene induction after focal ischemia have been reported, and these patterns may contribute to the delayed progression of damage or, alternatively, may mediate neuroprotection, tissue remodeling, and functional compensation.9 In recent years there have been an increasing number of proteomics studies investigating the alterations in protein expression relevant to human diseases, but few of them have focused on strokes. Nonetheless, up-regulations of dihydropyrimidinase-related protein 2, spectrin alpha II chain, heat-shock cognate protein 70 pseudogene 1, and tropomodulin 2 have been found via MS after focal cerebral ischemia.10
The goal of the present study was to determine the protein profile in rat brains following experimental stroke induced by transient unilateral occlusion of the middle cerebral arteries (tMCAO). Identifying proteins with altered expression following a stroke may provide potential targets for neuroprotective therapy that would be of benefit to stroke patients. Fifty-eight protein spots with differential expression levels were found. Seven differentially expressed proteins were identified by matrix-assisted laser desorption ionization (MALDI)-time-of-flight (TOF) MS. These findings provide new clues regarding the mechanisms underlying transient ischemia and reperfusion injuries in cortical neurons.
Methods
Animals
Adult Sprague-Dawley rats (n=12) were anesthetized using chloral hydrate (400 mg/kg, i.p). All procedures were approved by the Institutional Animal Care and Use Committee of the National Defense Medical Center, and were compliant with the guidelines of the National Science Council on Animal Care.
MCA ligation
Temporary ischemia was induced by clipping the bilateral common carotid artery (CCA) and ligating the right-side middle cerebral artery (MCA), as suggested by Chen et al.11 The bilateral CCA was isolated via a midline cervical incision, and then temporarily clipped with arterial clips. A right-side middle squamous craniotomy with a diameter of 2.0-3.0 mm was performed, and the MCA was ligated using a 10-O suture. The suture was then released after 60 min of ligation, and the craniotomy site was covered with Gelfoam and bone wax. Individual animals were perfused intracardially at 0, 6, or 24 h (n=3 animals per time point) after ligation for reperfusion protein assay.12
Sample preparation
Animals were anesthetized and decapitated, and their brains then microdissected on ice under a surgical microscope to initially retrieve the cerebrocortices. The harvested brain tissue was cut into several 500-µm-thick sections. Punch separation of the cerebrocortex sections was then performed. The tissues were homogenized on ice with a tissue tearer in 300 µL of sample buffer {58 mM dithiothreitol, 65 mmol/L 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonic, 7 M urea, 1.9 mol/L thiourea, 1.75% pH 3-10 carrier ampholytes}. The mixture was then mixed for 1 h and centrifuged at 18000×g for 10 min. Protein concentration was determined in the soluble supernatant using the RC-DC-Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA) to avoid interference from ampholytes and reducing agents present in the buffer.
2-DE
A 2-DE system was used to separate proteins obtained from the cerebrocortex samples (250 µg/sample). An isoelectric focus over a pH range from 3 to 10 and a second dimension slab gel were used to separate the proteins by molecular weight while a first dimension gel was loaded on a large format (22×22 cm). The gels were then stained using fluorescent SYPRO Ruby (Molecular Probes, Eugene, OR, USA) to quantify the proteins, and images were obtained using HT 2-D image analysis software (Genomic Solutions Inc., Ann Arbor, MI, USA).13
In-gel tryptic digestion
Samples were prepared using a modification of a previously described technique.14 Water (18 mol/L) was then used to wash the stained gel slabs extensively, excising the spot with a volume around 1-3 mm3. The gel pieces were mixed with ammonium bicarbonate (0.1 mol/L) to double the volume and then incubated at room temperature (20℃) for 15 min. The gel pieces were dried using a high-speed vacuum centrifuge after the solvent had been removed, rehydrated with 20 µL of 20 mmol/L dithiothreitol in 0.1 mol/L NH4HCO3, and then incubated at 56℃ for 45 min to reduce the protein. When the tube was cooled to room temperature, the dithiothreitol solution was removed before 20 µL of 55 mmol/L iodoacetamide in 0.1 mol/L NH4HCO3 was added, and the gel incubated at room temperature in the dark for 30 min, followed by further incubation for 15 min in 0.2 mL of 50 mmol/L NH4HCO3, before adding 0.2 mL of acetonitrile. At the end of another 15-min incubation period at room temperature, the solvent was removed and the gel pieces were dried in a vacuum centrifuge. The gel pieces were rehydrated with 5 µL of 20 ng/µL modified trypsin (Promega, Madison, WI, USA) in 50 mmol/L NH4HCO3 and then covered with 50 mmol/L NH4HCO3 solution and incubated overnight at 37℃.
Sample preparation for MALDI-TOF MS
A nitrocellulose solution was made by dissolving a nitrocellulose membrane in 1:1 acetone/isopropanol solvent. Alpha-cyano-4-hydroxycinnamic acid (-CN) was washed with 50 µL of acetone and the acetone phase was discarded. The -CN was dissolved in acetone to a concentration of 10 mg/mL, and then the nitrocellulose and -CN solutions were mixed at a ratio of 1:4; 1 µL of this mixture was deposited onto a 96-well MALDI target plate.
A sample was prepared for addition to the plate by adding 2 µL of sample to 2 µL of a solution of acetone-washed -CN dissolved in 0.1% trifluoroacetic acid and added to a 1:1 H2O/acetonitrile mix to a final -CN concentration of 10 mg/mL. An aliquot (1 µL) of the sample mixture was loaded onto each thin film. After the sample mixture was dried, 1.5 µL of 2% formic acid in 18-MΩ water was added to each spot. The formic solution was removed by gentle blotting. This washing step was performed twice. The samples were then dried at room temperature. Fragment sizes were determined by MALDI-TOF MS.
Analysis of peptide sequences
Protein identification from tryptic fragment sizes was made using the Mascot search engine (www.matrixscience.com) based on the entire National Center for Biotechnology Information protein database under the assumptions that peptides are monoisotopic, oxidized at methionine residues, and carbamidomethylated at cysteine residues. Up to one missed trypsin cleavage was allowed, although most matches did not contain any missed cleavages. An accuracy of 100 ppm or greater for the tryptic sizes was required. The original protein size based on the portion of the gel from which the spot was excised was not restricted. All matches using mass values (either peptide masses or MS/MS fragment ion masses) were handled on a probabilistic basis. The total score is the probability that the observed match is a random event. The direct reporting of probabilities can be confusing, and so the probability-based molecular weight search score was used, defined as -10×log10 (p), where p is the absolute probability;15 thus, a probability of 10-20 becomes a score of 200. In this study, scores of >70 were considered significant (p<0.05), and all protein identifications were in the expected size range based on their position in the gel.
Identification of differentially expressed proteins
Silver staining was performed using silver nitrate in combination with formaldehyde developer. The detection sensitivity of the silver stain was 0.5 ng. SYPRO-Ruby-stained gels were scanned using a high-resolution 12-bit camera and analyzed using HT 2-D gel software (Genomic Solutions). A composite gel was formed using gels obtained from each treatment. Bioimage software was initially used to identify matching spots in each tissue sample according to the manufacturer's instructions. The accuracy of the protein-spot matching was determined manually for each spot on each gel. The spot intensity was compared for each protein identified and matched, and the integrated intensity of each spot was determined for each of the eight gels. Except where indicated otherwise, the data are presented as mean±SEM values, and a Mann-Whitney test was carried out on each to determine the significance of any differences.
Results
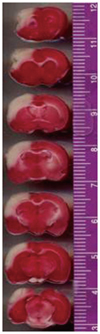
The true infarct volume obtained 24 h after tMCAO was 306.17 mm3 (n=3). Infarcted tissues were found predominantly on the lesioned sides of the cortices (Fig. 1). No infarctions were observed on the nonlesioned sides or in the sham-operated controls.
Two-dimensional gel electrophoresis maps were constructed for rat cortical proteins with or without tMCAO at pH 3-10. A typical 2-DE map of each group obtained from different cortical areas of the brain is shown in Fig. 2. After silver staining, about 400 spots in total were resolved in both maps and compared. There were no significant differences between the groups.
In addition to silver staining, SYPRO Ruby protein gel staining was performed to investigate the protein profile in the rat brain during the reperfusion stage following tMCAO. The 2-DE maps of sham-operated animals were compared at 6 and 24 h after tMCAO (Fig. 3). About 400 spots were found and histograms of each spot at different time points (0, 6, and 24 h following tMCAO) were compared (Fig. 4). The silver stain did not reveal significant up- or down-regulation of the protein profile, but the SYPRO Ruby stain revealed 8 spots of down-regulation and 39 spots of up-regulation at 24 h after reperfusion. On the other hand, 11 spots were up-regulated at 6 h and then returned to noninjury levels by 24 h. After in-gel digestion, the proteins were eluted from the spots and fingerprint patterns of peptides were prescribed by MALDI-TOF analysis. The proteins were identified after Mascot search comparison (Fig. 5). Of these, only seven were successfully identified by in-gel digestion by MALDI-TOF analysis, with a protein sequence coverage of 17-31% and top scores ranging from 62 to 111 (Table 1). Five spots were identified as aconitase 2, neurotensin-related peptide (NRP), hypothetical protein XP-212759, 60-kDa heat-shock protein (HSP60), and aldolase A. The expression levels of these proteins were up-regulated by 1.43-5 times in comparison to their baseline expressions in the histograms. The observed molecular masses differed from the theoretical values by no more than 5-6%, and the observed isoelectric point values were also in close agreement with the theoretical values.
Discussion
To the best of our knowledge this is the first published analysis of the spatial and temporal expression of proteins from the cerebral cortex after ischemic stroke. Approximately 400 protein spots were resolved by 2-DE, and compared with sham-group samples. The silver staining did not show significant up- or down-regulation of the protein profile, but the SYPRO Ruby stain revealed 8 spots of proteins that were down-regulated and 39 spots of proteins that were up-regulated at 24 h after the reperfusion. It is perhaps somewhat surprising that only seven spots were identified. However, this should be seen in the context of the analyses having been done at a different time points, namely 0, 6, and 24 h, after tMCAO. Of the seven protein spots that were identified, one was up-regulated at 6 h postischemia, five were up-regulated at 24 h postischemia, and one remained unidentified. The identified proteins are discussed individually below.
Protein up-regulation at 24 h after tMCAO
Aconitase 2
Aconitase is an iron-sulfur protein, or nonheme iron protein. Its iron-sulfur clusters are thought to play a role in the electron-transfer reactions of oxidative phosphorylation.16 The function of aconitase is to catalyze both steps so that isomerization of citrate is accomplished by a dehydration step followed by a hydration step in the citric acid cycle. The result is an interchange between a hydration atom and a hydroxyl group.
Mitochondria are the principal intracellular sources of superoxide (O2-) and hydrogen peroxide under both physiological and pathological conditions, and are therefore primary loci for reactions in these species and those derived from within their cells.17,18,19 Mitochondrial aconitase (m-aconitase), an enzyme that catalyzes the reversible isomerization of citrate and isocitrate via cis-aconitate in the Krebs cycle, contains a [4Fe-4S] prosthetic group in which one of the types of iron, Feα, is not ligated to a protein residue, and can thus bind to hydroxyl groups of substrates or water.20,21 M-aconitase is highly sensitive to O2--mediated inactivation; O2- selectively reacts at 107/M/s with the iron-sulfur cluster, leading to the release of Feα. M-aconitase is reactivated by the reincorporation of Fe2+, which is facilitated by reductants such as glutathione. The ratio between [4Fe-4S]-aconitase and [3Fe-4S]-aconitase has been used to calculate the steady-state concentration of O2- in cells.22
M-aconitase is a multifunctional protein: on the one hand it is a key enzyme for the Krebs cycle, while on the other hand it can act as a sensor in the redox regulation of metabolism by O2-.23 The exquisite sensitivity of m-aconitase to inactivation by O2- may provide a control mechanism whereby decreasing the level of reducing equivalents (i.e., NADH) entering the electron transport chain would regulate the level of O2- produced by mitochondria.23,24
Aconitase activity is also used in mitochondria activity assays. The mitochondrial activity in neurons increases to produce more ATP for cell repair-especially at the penumbra region-during the reperfusion state. On the other hand, increasing the level of O2- after transient ischemia and reperfusion periods was found to suppress the activity of aconitase.23 Aconitase may have created more for the energy requirements while their activity was being affected by higher levels of O2-. It can be speculated that the increase in aconitase enzyme levels found herein is due to increasing mitochondrial activity toward the production of more ATP for cell repair in the penumbra region.
NRP
Neurotensin is a tridecapeptide that is found within neurons in the brain and spinal cord, in endocrine cells in the pituitary gland and small intestine, and in chromaffin cells in the adrenal gland.25 It is reported to have a broad spectrum of biologic effects, including effects on the endocrine, cardiovascular, digestive, and reticuloendothelial systems, as well as on temperature regulation, nociception, and behavior. Blood levels of immunoreactive neurotensin increase dramatically with the ingestion of food, and particularly fats. NRP releases histamine from mast cells and increases cutaneous vascular permeability; therefore, it might be speculated that NRP (or similar peptides) functions as an inflammatory mediator. In this regard, NRP could be formed locally in a manner similar to bradykinin, perhaps via the release or activation of acid protease activity, such as that of cathepsin D, and the second phase of inflammation is thought to be initiated by the release of lysosomal enzymes from phagocytes, elevating the acid protease activity in the skin during an inflammatory response.26 The increases in NRP after the reperfusion period (24 h after tMCAO) in the present study may thus have been due to inflammation reactions during the reperfusion state.
Hypothetical protein XP-212759
Hypothetical protein XP-212759 is an unknown protein that was identified in the present analysis. While bioinformatics tools have progressed remarkably, providing biologists with valuable information for functional elucidation, the prediction of protein function from sequences and structures remains difficult because homologous proteins often have different functions.27 The existence of hypothetical proteins in genomes constitutes a major issue for comparative and functional genomics analyses. In particular for pathological conditions, these hypothetical proteins hamper the search for new and effective drug targets, and weaken progress in researching conditions such as strokes, rendering it difficult to improve our understanding of pathogenicity.28
HSP60
The levels of HSP60 increased by around 3.5-fold at 24 h after ischemia in the penumbra region in this study; this protein was detected in both the cytoplasm and mitochondria. Purified cytoplasmic HSP60 exhibits chaperone activity, and the protein is imported into the mitochondria in vitro via a mitochondrial import assay. Under normal conditions, mammalian HSP60 is located both in the cytoplasm (as stable cytoplasmic HSP60) and in the mitochondria. Cytoplasmic HSP60 is rapidly imported into the mitochondria under severe conditions by cytoplasmic HSP70.29 HSP60 and HSP10 are stress-inducible mitochondrial matrix proteins that form a chaperonin complex that is important for the folding and assembly of mitochondrial proteins. Simultaneous induction of the modified RNAs (mRNAs) for the mitochondrial chaperonins HSP60 and HSP10 in various regions in focal cerebral ischemia demonstrates that mitochondrial stress conditions persist concomitantly with cytosolic stress conditions in this state.30 Furthermore, apoptosis is closely linked with mitochondrial dysfunction, which is involved in delayed neuronal death following brief periods of global cerebral ischemia,31 and this could be the reason why the protein was up-regulated at 24 h after the ischemia in the penumbra region.
Aldolase
Aldolase, an enzyme of the glycolytic pathway that catalyzes the aldol cleavage of fructose-1,6-biphosphate into glycerinaldehyde-3-phophate, was increased in the penumbra region by about fivefold in the present study at 24 h after ischemia. This enzyme is involved in the breakdown of glucose, fructose, and galactose, a process used by cells to generate energy in the form of ATP. Cell death after a stroke involves apoptotic, autophagocytic, and necrotic mechanisms that may cause the release of cytosolic proteins into the extracellular space. Release of aldolase into the extracellular space of the central nervous system has been found to take place in various disease states and in the presence of brain injury.32 The release of aldolase into the cerebrospinal fluid after stroke in vivo, as well as into the extracellular space after hypoxia in cell culture, has also been demonstrated.33
Up-regulation of dynamin-1 at 6 h after reperfusion
Levels of dynamin-1 increased by about 2.2-fold at 6 h after ischemia in the penumbra region. The DTPase dynamin is required for late-stage endocytic clathrin-coated vesicle formation. Dynamin is targeted to coated pits on the plasma membrane in a guanine-nucleotide-independent manner, through interactions between its praline, arginine-rich COOH-terminal domain and the SH3-domain-containing protein amphiphysin. Mammals express three dynamins (dynamin-1, -2, and -3) with different expression patterns.34 Dynamin-1 is expressed exclusively in the brain35 and in neurons, and levels of dynamin-1 increase with synapse formation in parallel with the levels of synaptic vesicle proteins.36 Furthermore, dynamin-1 plays a dedicated and essential role in the recycling of synaptic vesicles, which is critical to nervous system function.37,38 A late depolarizing postsynaptic potential induced by hippocampal CA1 pyramidal neurons after transient ischemia indicates an enhancement of synaptic transmission following ischemia.39 The present data are compatible with those of reports indicating that synapse-releasing efficacy is increased during reperfusion periods.39,40
Down-regulation of glucose-regulated protein 78-kDa (GRP78) at 24 h after reperfusion
The glucose-related stress response is part of a general cellular defense mechanism, referred to as the unfolded protein response (UPR), which is induced by glucose or oxygen starvation.41,42 One characteristic of the UPR is the induction of endoplasmic reticulum (ER) resident stress proteins, referred to as the glucose-regulated proteins (GRPs), which are Ca2+-binding chaperone proteins with protective properties.43 The glucose-regulated protein GRP78, a 78-kDa protein (also referred to as binding immunoglobulin protein), is one of the best-characterized GRPs and is known to form complexes with heterologous proteins that are processed through the ER.44 GRP78 can bind to malfolded proteins and unassembled complexes, and can protect cells against cell death caused by disturbances of the ER homeostasis.45,46,47,48 GRP78 has also been induced in endothelial cells damaged by reductive stress caused by hyperhomocysteinemia, which is a common risk factor for thrombotic vascular events such as premature arteriosclerosis, strokes, myocardial infarctions, and thrombosis.49,50 Therefore, the induction of GRP could be an adaptive response that has evolved in mammals to protect endothelial cells against stress-induced death. GRPs are induced in response to stress, but once the stress is removed these proteins are posttranscriptionally modified into biologically inactive forms.44 GRP78 is critical for the maintenance of cell homeostasis and the prevention of apoptosis. The present data show that this protein is present at very high levels at the beginning of the reperfusion period, or 1-6 h after tMCAO; however, as shown in Fig. 4B, it was relatively down-regulated at 24 h after reperfusion on the lesioned sides. This is probably the result of cell loss on the lesioned side relative to the contralateral side during the ischemic and reperfusion periods. In addition, the down-regulation of this molecular chaperone by tMCAO may indicate the potential for it to impact immune function on multiple levels.
Limitations and correlations
Matrix-assisted laser desorption ionization-time-of-flight mass spectometry was used to evaluate the protein expression at various times after tMCAO. Sequence-similarity searches were conducted to enable proteomic analysis and comparison of proteins on the ischemic lesioned sides with those on the contralateral side at various time points in order to identify the protein changes during the postischemia period. The proteins that exhibited significant fluctuations included 1) the functional mitochondrial proteins, including HSP60 and aconitase; 2) NRP, the protein related to inflammation; 3) proteins associated with metabolism, including aldolase C and GRP78; 4) dynamin-1, which is involved in the release of synaptic neurotransmitters; and 5) one unknown hypothetical protein. These protein-level changes indicate the occurrence of changes in the systems involved in postischemic functional changes in the penumbra area.
There are also similar studies of brain ischemia that have described the following important protein level changes. First, functional changes in the pathways involving mitogen-activated protein and AKT kinase activity reflect the associated vulnerability to cerebrovascular injury. The alteration of gene and protein expression patterns, as well as the phosphorylation state of adenylyl-cyclase-associated protein 2, the so-called Turned On After Division-64 kDa protein, propionyl CoA carboxylase, APG-1, and valosin-containing protein (the kinase target) are consistent with the increased vulnerability of specific strains of animal (e.g., the spontaneously hypertensive stroke-prone rat) to cerebrovascular injury.51 Furthermore, stroke susceptibility was associated independently with multiple protein changes associated with ischemia, angiogenesis, or blood-brain barrier integrity. Aquaporin-4 and laminin-alpha-1 induction in cerebral cortical microvessels is associated with stroke susceptibility. Significant molecular changes in ischemic cerebral microvasculature in the prestroke stage could contribute to the observed model phenotype of microhemorrhages and postischemic hemorrhagic transformation.52 Moreover, six plasma proteins, comprising alpha-2-macroglobulin, complement C3, inter-alpha-trypsin inhibitor heavy chain H3, serum albumin, haptoglobin, and transthyretin, which are classified as acute-phase proteins, changed significantly at 24-h postreperfusion after 90 min of left-MCA occlusion.53 In addition, the analysis of the rat proteome in a study of the intestinal mucosa revealed that those proteins involved in the cellular processes of energy metabolism, antioxidation, and antiapoptosis changed by more than 1.5-fold following intestinal ischemia and reperfusion. Within these proteins, aldose reductase (AR) could remove reactive oxygen species, indicating that AR may play a key role in intestinal ischemic protection.54
However, the data obtained in the present study were somewhat discrepant from those obtain in previous studies, which may have be due to the actual levels of verified proteins being lower than had been expected and because of poor sequence representations for proteomic identification by theoretical translation of publicly available data.55 Overcoming the weakness of sequence-similarity searches requires consideration of several "similarity-free" methods,56 including structural similarity searches looking for the global folding of the protein57,58,59,60 or detecting the functionally important regions of the protein.61,62,63,64 Thus, further experimentation is necessary to confirm the exact assignment of functions to the unknown protein.





 XML Download
XML Download