

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Lipoid proteinosis (LP; OMIM 247100), also known as Urbach-Wiethe disease, is a rare autosomal recessive disorder characterized by variable scarring and infiltration of the skin and mucosa. Clinical features include warty skin infiltration, papules on the eyelids, and skin scarring, in addition to extracutaneous abnormalities such as hoarseness of the voice, epilepsy, and neuropsychiatric abnormalities.1 In severe cases this infiltration can lead to respiratory obstruction. Histological examinations reveal the widespread deposition of hyaline material and disruption of the basement membrane around blood vessels and at the dermal-epidermal junction.
The molecular basis of LP has been elucidated: it appears to result from mutations in the gene encoding extracellular matrix protein 1 (Ecm1, ECM1; OMIM 602201).2 Ecm1 is a key protein in epidermal differentiation, binding of dermal collagens and proteoglycans, and regulation of angiogenesis. Homozygous and compound heterozygous mutations in ECM1 have previously been described in patients with LP. ECM1 maps to chromosome 1q21.2, and there are three known splice variants (Ecm1a, Ecm1b, and Ecm1c) and a putative fourth variant.2,3 Ecm1a is the most widely expressed splice variant, while Ecm1b has a much more restricted expression pattern.4 Ecm1 contains numerous cysteine residues that are arranged in a specific manner. The cysteine-containing domains all have the typical CC-(X7-10)-C arrangement that is capable of the forming protein double loops involved in protein-protein interactions.5 Therefore, Ecm1 and its alternative splicing isoforms may serve as binding and transport proteins in epidermal differentiation.6 Ecm1 was found to negatively regulate endochondral bone formation by inhibiting alkaline phosphatase activity and mineralization.7,8
This report describes the case of an LP patient who carries a homozygous, undescribed nucleotide transition in exon 7 of ECM1 causing a homozygous nonsense mutation. Her clinical, histological, and molecular features indicate that the truncating mutation of exon 7 causes a severe phenotype.
Case Report
Patients
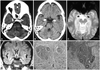
The index patient was a 35-year-old woman born to unrelated parents who developed a progressively hoarse voice from 9 months of age, and vesicular and bullous lesions that appeared after minimal trauma. A physical examination revealed deposition of hyaline material in the larynx, epidermolysis bullosa, multiple yellowish warty skin papules on the body, and diffuse acneiform scars over her face, with blisters in the mouth, eyes, and areas subject to friction. Neurological symptoms included unilateral paresthesia, dizziness, and memory loss associated with depression, in addition to a decline in verbal fluency and impaired coordination of movement from the age of 19 years. Computed tomography revealed bilateral and symmetric amygdaloid-uncal calcifications (Fig. 1A and B). Gradient refocused-echo T2*-weighted MRI images (Fig. 1C and D) revealed marked hypointense bilateral rounded symmetric lesions in the mesial temporal lobes, involving the amygdaloid complex. A routine EEG (awake and sleep) recording was slightly unstable, but no epileptiform activity was evident. A cognitive event-related potential P-300 wave revealed a reaction-time delay during development of the Posner task, with a normal latency of the P3 component during the oddball task. The results of a neurological examination were unremarkable, and a neuropsychological evaluation revealed a decrease in information processing speed and disturbances in episodic memory and the amnesic evocation process. These findings could be explained by her depressed mood. The results of a blood workup were unremarkable.
A skin biopsy (Fig. 1E and F) of the granuloma annulare revealed sweat glands and small nerves with deposition of strongly periodic-acid-Schiff-positive homogeneous, eosinophilic, hyaline-like material and diastase resistant granules around dermal blood vessels, revealing its glycoproteic nature, which is consistent with LP.
Sequencing of the coding region of ECM1
Genomic DNA was purified from blood samples using a salting-out procedure, and stored in TE buffer. ECM1 was amplified by a polymerase chain reaction (PCR) with primers spanning all ten exons and intronic flanking regions. Genomic DNA (100 ng) was amplified in 20-µL reaction volumes containing 20 pmol of primers (Table 1), 1.5 nmol of MgCl2, 4 nmol of dNTP, and 1 U of AmpliTaq DNA polymerase (Applied Biosystems, Warrington, UK). The PCR products were separated by 1.5%-agarose-gel electrophoresis and purified with the Qiaquick Gel Extraction Kit (Qiagen, Crawley, UK). Amplified fragments were run and sequenced in an ABI 3130 genetic analyzer.
Sequencing of exon 7 revealed the presence of a single base substitution at position 1076 of cDNA (GenBank accession no. NM_004425.3). The c.1076G>A transversion causes a premature termination codon, p.Trp359*, that affects the Ecm1a isoform. The patient was found to be homozygous for the mutation, while her parents were heterozygous carriers (Fig. 2). No other changes or polymorphisms were found in the ECM1 coding region. The mutation causes the loss of a restriction target for NlaIII endonuclease, which was therefore used as a screening test. The mutation was not present in 106 chromosomes of unrelated control subjects.
Haplotype study
Four different polymorphic dinucleotide microsatellites were analyzed in this study: D1S2344, D1S2345, D1S305, and D1S2624. The primer sequences were obtained from the uniSTS database of the National Center for Biotechnology Information. The sizes of the microsatellites were determined by direct sequencing in the ABI 3130 genetic analyzer.
The four markers allowed determination of the segregation phase and the haplotypes associated with the c.1076G>A transversion. The parents' haplotypes differed significantly in the mutated chromosomes, although double recombination events cannot be ruled out (Fig. 2).
Discussion
Lipoid proteinosis was first described in 1929, but the current literature still contains only around 300 published cases. A Spanish patient carrier of a homozygous mutation (c.1076G>A) ECM1 has been described herein. A previous report5 described a different mutation in two Caucasian brothers (c.1077G>A). Nonetheless, the same stop codon, W359X, predicts a truncated protein in both cases. Reports on the genotype-phenotype relationship point to a milder phenotype in carriers of missense mutations in the Ecm1a isoform, whereas mutations in the Ecm1b isoform (i.e., with exon-7 skipping) have been associated with more severe phenotypes.9 This is in agreement with the present finding in a Spanish patient carrying a truncating mutation in exon 7 and exhibiting a complete dermatological and neurological manifestation. It is difficult to study the genotype-phenotype correlation because of the different ages of patients, since many clinical features vary with age. The patient studied herein reported spontaneous hemorrhagic blisters under both humid and very dry environmental conditions, which suggests that both genetic and environmental factors can modify the disease severity. Therefore, genotype-phenotype differences among reports can be at least partially attributed to age and nongenetic factors that differentially affect the development of neurologic symptoms and the severity of this disease.
Extracellular matrix protein 1 has specific binding sites for several extracellular matrix proteins and polysaccharides: 1) the NH2-terminal free cysteine domain can bind collagen IV [amino acids (aa) 14-207]; 2) the tandem repeat can bind fibulin 1C/1D, matrix metalloproteinase 9 (MMP-9), and laminin (aa 207-361); 3) the C-terminal region can interact with domain V of perlecan (aa 424-540); and 4) Ecm1 promotes the link between collagen IV and laminin.6,10-12 Moreover, Sercu et al.12 suggested that Ecm1 can act as a "biological glue" in the skin's basement membrane to maintain the structural and functional integrity of this organ. If translated, the stop codon described in the present Spanish patient predicts a truncated protein that would impair the interaction with perlecan, MMP-9, fibulin, and laminin. Given the lack of symptoms in the carrier parents, haploinsufficiency or toxic effects could be discarded as pathogenic mechanisms. Extracutaneous manifestations such as epilepsy and neuropsychiatric disorders13,14 can be related to the inhibition by Ecm1 of MMP-9 activity,10 a protein that is strongly expressed in the brain.15 The truncated 358 polypeptide lacks regulatory MMP-9 activity and the aforementioned binding domains to several other matrix components, and therefore causes impairment of the extracellular matrix structure. The histological images obtained in this study clearly showed material deposition around sweat coils and capillaries.
A new mutation in exon 7 of ECM1 is described herein that leads to a premature termination codon as the cause of a severe phenotype of LP. The lack of coincidence in the parents' haplotypes is noteworthy, although a recombination among the microsatellite markers cannot be ruled out. In conclusion, the identification of pathogenic mutations in ECM1 is significant for genetic counseling with respect to clinical severity and prognosis.


 XML Download
XML Download