

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Reversible posterior leukoencephalopathy syndrome (RPLS), also referred to as posterior reversible encephalopathy syndrome, typically presents with headache, altered mental status, visual disturbances, and seizures in the setting of acute blood pressure alterations.1 However, it has also been associated with sepsis, shock, posttransplantation, postchemotherapy, and medications such as cyclosporine and tacrolimus in the absence of blood pressure changes.2 Radiologically, RPLS is characterized by symmetrical subcortical areas of vasogenic edema that are preferentially parieto-occipital, and it typically resolves within a few weeks after appropriate treatment.1,3,4
The term RPLS may not cover the complete spectrum of the condition, since nonreversible lesions are also reported, as well as lesions in other parts of the brain including the gray matter.3,5 As another example of nontypical RPLS we present a patient with unilateral RPLS that developed 21 days after coiling of an intracranial aneurysm.
Case Report
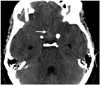
A 36-year-old woman was admitted with acute severe headache. She had also experienced an epileptic seizure and had a slightly lowered consciousness at admission. Nonenhanced computed tomography (CT) showed subtle signs of subarachnoid hemorrhage (SAH) with some frontobasal subarachnoid blood (Fig. 1). CT angiography revealed a 4-mm aneurysm of the left anterior cerebral artery (ACA). At admission her blood pressure was 128/55 mm Hg. On the first day of admission her mean arterial pressure (MAP) varied between 81 and 99 mm Hg, with a mean of 88 mm Hg. She had no history of hypertension, was no taking any medication, and exhibited a good neurological condition (Hunt and Hess grade of 1).
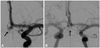
The aneurysm was coiled on the same day. The procedure took longer than usual (2 hours) because of the smallness of the aneurysm, which made it difficult to enter it safely with the microcatheter. During the procedure, both internal carotid arteries were catheterized to optimize depiction of the local vascular anatomy (Fig. 2). However, a microcatheter was only advanced in the left ACA to reach the aneurysm. According to the local protocol, 5000 IE of heparin was given during the procedure; no other medication was given during the procedure. The aneurysm was successfully packed with two small coils (3 cm long; 2-mm UltraSoft, Boston Scientific, Natick, MA, USA) using a left approach. No complications were noted and the patient experienced no focal neurological symptoms.
After the procedure the patient developed periods of bradycardia with sinus arrest in the intensive care unit (ICU) but without clinical symptoms. There were assumed to be secondary to the SAH, and were treated conservatively. Transcranial Doppler (TCD) performed during the following week showed mildly increased flow velocities in the middle cerebral artery (MCA) that were just within the normal range (111 and 82 cm/s in the left and right MCAs, respectively), suggesting mild vasospasm. Oral nimodipine at 30 mg taken 12 times daily and triple-H therapy were started according to the local protocol. On day 4 of the triple-H therapy the MAP reached 72-108 mm Hg (mean 93 mm Hg). Slight lung edema was treated with furosemide. TCD performed 5 days later showed normal MCA velocities, although the velocities in the MCA and ACA continued to be higher on the left than on the right, albeit still within the normal range. Triple-H therapy was subsequently stopped. During the hospital stay the clinical status of the patient improved and her headaches gradually diminished. The patient was discharged in good clinical condition after 12 days with a prescription for oral nimodipine (60 mg taken six times daily for 20 days). The MAP at discharge was 100 mm Hg.
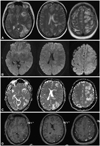
The evening after taking the last prescription of nimodipine, which was 21 days after coiling and 15 days after the cessation of triple-H therapy, the patient presented at the emergency room with progressive headache, right-sided homonymous hemianopsia, right-sided tactile extinction, and a slight paresis of the right arm. Her MAP during the first day of re-admission was 81-120 mm Hg (mean 108 mm Hg). The cerebrospinal fluid pressure measured via a lumbar puncture was elevated at 42 cmH20. Spinal fluid showed residual signs of the recent SAH but no signs of meningitis. Renal function was normal with serum creatinine, sodium, and potassium levels of 47 µmol/L, 140 mmol/L, and 4.4 mmol/L, respectively. Nonenhanced CT performed 21 days after the first onset of the SAH revealed subcortical hypodense areas in the left hemisphere occipitally and around the insula. Magnetic resonance imaging (MRI) showed vasogenic edema mostly subcortically in the entire left hemisphere, with a preference for the watershed areas (Fig. 3). No susceptibility artifacts were seen within the parenchyma. Diffusion-weighted MRI images showed no signs of ischemia. After gadolinium administration there was patchy enhancement in the lesions (Fig. 3). CT angiography did not show vasospasms or vascular occlusions.
The combination of headache, confusion, visual disturbances, and vasogenic edema on MRI was suggestive of RPLS. Further clinical analysis revealed no signs of infection, and dural sinus thrombosis was ruled out with CT venography. Because late-onset secondary brain ischemia was initially suspected clinically, oral nimodipine was restarted at a daily dose of 180 mg. This improved the clinical status of the patient, with headache being the most prominent symptom; this was reduced by prescription of dexamethasone at 2 mg twice daily. Slow tapering of dexamethasone led to the return of slight headaches, but they could now be controlled with acetaminophen. Because the initial clinical suspicion of ischemia was ruled out by the imaging workup, a diagnosis of RPLS was eventually suggested on the fifth day of re-admission based on clinical signs and MRI findings. By the sixth day of re-admission the MAP had decreased to 82-100 mm Hg (mean 89 mm Hg), possibly due to the nimodipine treatment, and so no antihypertensive medication was administered in our patient. The patient was discharged and nimodipine was gradually reduced.
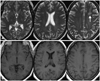
At three months after SAH the patient had no neurological symptoms apart from slight balance problems. The MRI abnormalities had diminished but were not completely resolved. However, MRI performed 7 months after the first presentation showed further reduction of the lesions, with a single small area of signal enhancement remaining in the left frontal lobe (Fig. 4).
Discussion
RPLS is generally thought to be associated with a sudden increase in blood pressure, which may exceed the cerebral autoregulation capacity and thus cause vasogenic edema in the brain.1 A second theory has also emerged after reports of RPLS combined with regions of vasospasm,5 in which hypoperfusion is assumed to cause injury and subsequent edema.
In general, etiological causes reported in cases of RPLS include eclampsia, chemotherapy, and systemic diseases such as systemic lupus erythematosus and Wegener's disease. RPLS has also been reported in patients after carotid endarterectomy (CEA).6,7 Because the brain perfusion is usually chronically low prior to a CEA procedure in patients with carotid stenosis, the restoration of 'normal' blood pressure may result in the autoregulation capacity being exceeded, thus provoking RPLS. Unilateral RPLS similar to our case has been reported8 in a patient after SAH and aneurysm clipping. In that case the unilateral distribution might have been due to treatment of combined carotid stenosis on the same side as the RPLS. Another report of unilateral RPLS9 has described a relation with cyclosporine. However, what distinguishes our patient from both of these previous cases is that she had no history of chronic hypoperfusion in the brain and was not on any long-term medication, which makes the unilateral distribution difficult to explain.
Bilateral RPLS has been described after coiling or clipping.10-13 Giraldo et al.10 suspected that this phenomenon was associated with hemodynamic augmentation (triple-H therapy) for symptomatic cerebral vasospasm after SAH. Some of the reported patients received hemodynamic augmentation therapy, despite having normal TCD results, which may have been the cause of their RPLS. However, unilateral edema was not seen in those patients.
A case report was recently published concerning a patient developing asymmetrical RPLS after SAH, clipping, two angiography procedures, and triple-H therapy.14 The distribution of RPLS was asymmetrical rather than being totally unilateral, and MRI showed characteristic bilateral RPLS lesions in the posterior lobes of the brain. Consistent with Giraldo et al.,10 hemodynamic augmentation therapy to treat vasospasm was suggested as the underlying cause.
Our patient also received triple-H therapy for a few days because of suspected vasospasm. However, her RPLS symptoms started 15 days after the cessation of triple-H therapy. This is a long period given that RPLS usually occurs subacutely.
Analysis of the time course of our patient revealed that only the cessation of nimodipine coincided with the onset of RPLS. Based on the vasoconstriction theory, it is possible that the acute cessation of nimodipine caused vasoconstriction. The left hemisphere previously showed slight vasospasm immediately after SAH and might have been more sensitive to changes in perfusion than the right hemisphere, which could explain the unilateral distribution. The restarting of nimodipine was associated with a reduction of headaches and neurological symptoms, and no symptoms returned when nimodipine was slowly tapered thereafter. However, we found no reports in the literature suggesting a causative relationship between the cessation of oral nimodipine and RPLS. An alternative explanation is that the elevated MAP at re-admission combined with the new clinical symptoms was associated with the brain abnormalities. Such blood pressure elevations may be treated with intravenous labetalol until they decrease by 25% or a diastolic pressure of 110.120 mm Hg is achieved. However, this was unnecessary in our patient since the MAP decreased within a few days of re-admission, possibly because of nimodipine treatment.
In summary, we have presented a case of unilateral RPLS related to SAH, endovascular aneurysm treatment, and vasospasm treatment. The striking asymmetry has raised several questions about the etiology of this phenomenon. Although we can only speculate about the underlying cause in our patient, the presented results emphasize the importance of distinguishing RPLS radiologically from other conditions such as ischemia in order to avoid inadequate treatment.
 XML Download
XML Download