

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Hereditary neuropathy with liability to pressure palsy (HNPP) is an autosomal dominant demyelinating peripheral neuropathy. The classical clinical presentation consists of recurrent, transitory, and painless focal neuropathies, mostly at sites of peripheral nerve entrapment and often triggered by minor nerve trauma.1 Anatomopathological examinations reveal segmental demyelination and remyelination with focally thickened myelin sheaths called tomacula1. HNPP is probably underdiagnosed, and reportedly 10-15% of mutation carriers remain clinically asymptomatic.2
Schwannomas, which are the most common benign peripheral nerve tumors in adults, are highly homogeneous tumors that are composed of Schwann cells and usually occur singly in otherwise normal individuals. Multiple schwannomas in the same individual suggest the existence of rare genetic diseases, such as neurofibromatosis type 2 (NF2), which is characterized by bilateral vestibular schwannomas. The occurrence of multiple nonvestibular schwannomas has been distinguished as schwannomatosis since it is a clinical entity distinct from other forms of neurofibromatosis.3
Hereditary neuropathy with liability to pressure palsy was diagnosed in one patient at the age of 48 years, and in his two sons, at the age of 8 and 16 years. Molecular genetic analysis revealed the usual 1.5-megabase deletion of the gene encoding peripheral myelin protein 22 (PMP22) on chromosome 17p11.2-12 in these three patients. The patient's father suffered from a sensory-predominant neuropathy, but he did not carry deletion of PMP22. His mother suffered from transitional numbness and palsy, but did not undergo PMP22 analysis.
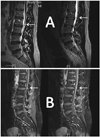
At the age of 52 years this patient presented with a voluminous and painful schwannoma of the left median nerve at the forearm, which required surgery; 2 years later lumbar magnetic resonance imaging (MRI) was performed due to chronic distal numbness in his lower limbs and intermittent pain in his left foot. A homogeneous, well-demarcated, and contrast-enhanced intradural ovoid mass (6×3×4.5 mm) was found localized to the left side, in front of the L3-L4 disc (Fig. 1). This mass was suggestive of a new schwannoma. The same patient presented 6 months later with a painful tumefaction at the lateral side of his right thumb. This tumor was removed and histopathological examination diagnosed a new schwannoma. Brain MRI revealed neither vestibular schwannomas nor meningiomas. Ophthalmological and dermatological examination yielded normal findings.
To the best of our knowledge, two other cases of genetically diagnosed HNPP combined with schwannomas have been reported.4,5 This third case strongly suggests that this association is not fortuitous. An association between schwannomas and Charcot-Marie-Tooth disease type 1A has also been reported.6
These data suggest that abnormal PMP22 status might predispose patients to the formation of schwannomas. The role of PMP22 is not fully understood. It is an integral membrane protein that is a major component of myelin in the peripheral nervous system, and is localized strictly to compact myelin. PMP22 mRNA is produced predominantly by myelinating Schwann cells.7 In HNPP, myelin with normal morphology is synthesized by Schwann cells, and deteriorates, particularly in areas subjected to trauma. PMP22 may be involved in the development and differentiation of Schwann cells, and may later act as a structural protein in myelin, controlling its thickness and stability. The role of PMP22 seems to be more important in the structural organization of the myelin than in the phenomena that precede its formation.8 Mutation of PMP22 may disturb the Schwann cell life cycle, thus potentially promoting the development of Schwann cell neoplasms.
Another hypothesis with respect to our patient is the existence of an association between HNPP and neurofibromatosis. Although genetic analysis for NF2 disease had not been performed, the presence of this condition was unlikely in our patient. Indeed, there was no evidence of vestibular tumors on high-quality cerebral MRI and no family history of NF2. However, this patient fulfilled the diagnosis criteria for schwannomatosis, which is a recently described entity characterized by the occurrence of multiple nonvestibular schwannomas.3
Most cases of schwannomatosis are sporadic. A study of schwannomatosis-related schwannomas has demonstrated the absence of the protein neurofibromin 2 and a multitude of truncating mutations of the gene encoding it (NF2), without a shared constitutional NF2 mutation in the different schwannomas of the same patient.9 NF2 is a tumor-suppressor gene located on chromosome 22. A germ-line mutation was reported in a case of familial schwannomatosis in the INI1/SMARCB1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1) gene, which is another tumor-suppressor gene located on chromosome 22.10 Since PMP22 encodes a protein that plays a role in the regulation of Schwann cell proliferation and life cycle, PMP22 mutations might lead to somatic instability mutations by inducing mutations in other tumor-suppressor genes, such as NF2 or INI1, and thus might predispose patients to the development of Schwann cell neoplasms.
The observed association between HNPP and multiple schwannomas is probably not fortuitous, and suggests a common genetic basis.
 XML Download
XML Download