

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Subacute sclerosing panencephalitis (SSPE), is a delayed, slowly progressive, and fatal form of encephalitis that occurs 6-10 years after measles infection, and normally progresses over a period of 12-18 months.1 Fulminant presentations with fatality within 6 months of symptom onset are very rare.2 Diagnosis in such cases may be difficult because the typical clinical features of reduced scholastic performance, cognitive impairment, myoclonus, and electroencephalogram (EEG) changes may not be seen initially. Atypical features such as acute vision loss, dysarthria, ataxia, focal and generalized seizures, epilepsia partialis continua, acute encephalitis, focal deficit, and asymmetric myoclonus have been noted.2,3,4,5,6,7,8,9,10 EEGs may also be atypical, showing periodic lateralized epileptiform discharges.10 We report herein a patient manifesting initially with ataxia, right hemiparesis, and asymmetrical delta-range slowing on EEG prior to the onset of Rademecker complexes, which are pathognomonic of SSPE. This case illustrates that a clinical picture suggestive of acute focal encephalitis could actually be a manifestation of fulminant SSPE.
Case Report
A 15-year-old boy presented with sudden-onset, nondisabling gait ataxia of 1 month duration followed by right hemiparesis, aphasia, and drowsiness of 2 days duration. He had no other neuraxial or constitutional symptoms. His vaccination status was erratic and he had a history of viral exanthematous fever at the age of 2.5 years.
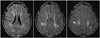
On admission he was drowsy and afebrile with normal vital signs. The findings of a systemic examination were unremarkable. On repeated commands, he attempted to answer questions with only one or two words. He performed single-step commands. His fundi and cranial nerve findings were normal. He had right hemiplegia (Medical Research Council Grade 2) with normal reflexes and extensor plantar response. He had mild choreoathetoid movements of the distal left extremities. In view of the multiaxial involvement and acute presentation, possible diagnoses of acute disseminated encephalomyelitis (ADEM) and viral encephalitis were considered. An EEG disclosed polymorphic, asynchronous 3- to 4-Hz delta activity over the entire left hemisphere, with normal background activity over the right hemisphere (Fig. 1A). Brain magnetic resonance imaging disclosed bilateral, subcortical, and posterior periventricular hyperintensities on T2-weighted, gadolinium-nonenhanced images (Fig. 2). All blood parameters were normal. His cerebrospinal fluid (CSF) was acellular with normal glucose and mild elevation in protein (60 mg/dL).
The presence of white-matter hyperintensities and albuminocytological dissociation on CSF analysis supported a diagnosis of ADEM. Intravenous methylprednisolone was administered at a dose of 1 g/day for the following 3 days, but had no impact-the patient deteriorated to a stuporous state. On the 4th day he exhibited slow myoclonic jerks involving the left-side extremities. A repeat EEG disclosed long-interval, high-amplitude, generalized periodic complexes of slow waves, lasting 1 second, with a slow background of low-amplitude, 3- to 4-Hz delta activity between them (Fig. 1B). Elevated titer of IgG antimeasles antibodies was detected in the CSF, and the CSF: serum ratio was 3. Amantadine and isoprinosine were started along with antiepileptics; However, the disease progressed relentlessly, and on the 7th day of hospitalization the patient became comatose and required ventilator support. He succumbed on the 13th day of hospitalization (i.e., 45 days from the onset of the illness).
After obtaining consent from the parents, a brain autopsy was performed within 2 hours of death. The brain was diffusely edematous with haziness of cortical and basal meninges. On serial coronal slicing, the white matter in the occipital, parietal, and temporal lobes had a granular gray appearance with areas of softening (Fig. 3A). The cortex appeared relatively normal, as did the deep nuclei of the basal ganglia and thalami. Gross sections through brainstem also exposed areas of softening and grayish discoloration in the midbrain (Fig. 3B). The cerebellum was well preserved.
Representative sections taken through different areas revealed predominant involvement of the bilateral visual, parietal, and temporal cortices. The sections revealed a diffuse encephalitic process with perivascular lymphocytic infiltration in the meninges, cortex, and white matter. The gray matter exhibited dense infiltrate of lymphocytes, histiocytes, and few plasma cells. There were large areas of neuronal loss with large reactive astrocytes highlighted by glial fibrillary acid protein immunostaining. The white matter from these areas also contained aggregates of histiocytes with demyelination. The neurons and the oligodendrocytes were loaded with smudgy intranuclear eosinophilic inclusions (Fig. 3C). The inclusions were strongly immunostained for the measles virus antigen (Fig. 3D). The midbrain also exhibited intranuclear inclusions and periaqueductal gliosis. Neither inclusions nor antigens were found in the cerebellum and cervical cord.
Discussion
Fulminant SSPE is a major diagnostic challenge because it can present with atypical features.2,3,4,5,6,7,8,9,10,11 Focal neurological deficits as an initial manifestation are very rare and may mislead the diagnosis.2,10,11 Our patient presented with ataxia and right hemiplegia and did not have any visual symptoms, seizures, or slow generalized myoclonus at the initial presentation. The asymmetrical slowing on the initial EEG only suggested a left hemispheric insult.
The characteristic slow generalized myoclonus that is characteristic of SSPE, and the periodic high-amplitude sharp and slow-wave discharges (Rademaker complexes) on EEG were initially absent in our patient. As noted in our patient and in previous reports,12 Dyken's clinical and EEG criteria are rarely fulfilled in patients with fulminant SSPE, and a high degree of suspicion is required to consider this possibility. The treatment options for SSPE are limited, and the outcome is fatal in almost all patients, although there are very rare cases of prolonged survival after administering intrathecal interferon, isoprinosine, amantadine, and immunomodulatory drugs.1
The histopathological picture may also differ from that noted in typical SSPE.12 Intranuclear inclusions were not easily demonstrated in some previously reported cases.12 Although the histopathology diagnosis of SSPE was clear in our patient due to the presence of inclusions and viral antigen in neurons and oligodendroglia, midbrain involvement was also seen, which is rare in typical SSPE. Rapid progression of the disease has been reported in SSPE patients with brainstem involvement.13,14 Brainstem involvement may thus determine the rapid decline of these patients.
Subacute sclerosing panencephalitis is a slowly progressive disease and normally does not manifest until years after the initial measles infection.1 The various mechanisms that have been implicated in the delayed manifestation include low virulence, mutated measles virus, and immunological defenses of the host.1,12 Paradoxically, fulminant SSPE has a rapid course after a long incubation period, and whether this suggests a sudden impairment of the host immune responses or an increase in virulence of the measles virus is not known. The various case reports in the literature suggest an association between the fulminant form of SSPE, lack of immunosuppression, and early onset of measles (occurring at less than 2 years of age).5 Similarly, our patient had not received a measles vaccination and had a history suggestive of measles infection early in his life. An etiological role of poor immune responses is also supported by a few reports of the sustained survival of patients with SSPE who received combination therapies that included intravenous immunoglobulins and isoprinosine.9,15 On the other hand, steroids have shown conflicting effects. Although most studies have shown that oral steroids may have no effect on the disease,15 in their case report, Serdaroglu et al.16 postulated that adrenocorticotrophic hormone may have worsened the clinical condition of their patient. The disease progressed rapidly after steroid therapy in our patient, which may have been due to a further impairment in immune responses or as a part of the natural course of the disease. Future reports may confirm or refute the negative impact of steroids on SSPE.
In conclusion, the present case increases the awareness of the fulminant presentation of SSPE and reiterates the atypical clinical, EEG, and histopathological findings associated with this form. Brainstem involvement, as proven by autopsy in our case, may determine the rapid decline of these patients.


 XML Download
XML Download