

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Inclusion-body myopathy (IBM) with Paget's disease of the bone (PDB) and frontotemporal dementia (FTD) (IBMPFD; Online Mendelian Inheritance in Man #167320) is a rare, late-onset autosomal dominant disorder arising from missense mutations in a gene on chromosome 9p21.1-p12 coding for valosin-containing protein (VCP).1 VCP, also known as p97, is a member of the ATPases associated with various cellular activities (AAA) family of proteins and is highly abundant, accounting for about 1% of total cellular protein in humans.2 This chaperone protein consists of two AAA domains (D1 and D2) that can bind and hydrolyze adenosine triphosphate. The obtained chemical energy is believed to be converted into mechanical forces that change the stereotactic conformation of the N-terminal domain. This N-domain (also known as cdc-40) can bind with a wide variety of tissue-specific cofactors and substrates, especially ubiquitin, and helps in the assembly, disassembly, or functional operation of protein complexes.2 As a consequence of its molecular promiscuity, mutations in VCP can hamper endoplasmic-reticulum-associated protein degradation, ubiquitin-related proteasomal protein degradation, the maturation of autophagosomes into autolysosomes, endosomal trafficking, the prevention of polyglutamin aggregation, the assembly of myosin, and the organization of myofibrils.2 These malfunctions account for what is probably only a slight imbalance in the complex processes controlling protein quality in cells, but they can lead to a long-term fatal accumulation of misfolded or aggregated proteins. The slowness of this intracellular accumulation would not only explain the late onset of the disease, but also its preferential phenotypic expression in organs with a low cell turnover, such as skeletal muscle and the central nervous system (CNS). At least 20 different missense mutations that can lead to a clinical picture of IBMPFD have been discovered, most of them in this N-domain.3
IBMPFD is believed to be underdiagnosed due to several factors that make it difficult to diagnose clinically.3,4 First of all, IBMPFD is a rare disorder, accounting for less than 1% of all patients with a familial form of FTD.5 Second, only a small group of all patients (12%) will ultimately exhibit the triad of cardinal symptoms that gave the disease its name: while 80-90% of all patients will have IBM and approximately half of them are diagnosed with PDB, only a third will develop ubiquitin-positive FTD.6 Moreover, the phenotypic expression of a mutation can vary so widely that carriers of the same mutation in a single affected family can have completely different symptoms: for example, one member might have PDB and FTD while his or her siblings might have only IBM.4 To complete the confusion, even two family members with IBM can exhibit a marked variability in the patterns of muscle weakness,7 or atypical features such as pyramidal tract dysfunction [mimicking spastic paraplegia or amyotrophic lateral sclerosis (ALS)], sensory motor axonal neuropathy, Parkinsonism, sensorineural hearing loss, cataract, cardiomyopathy, liver steatosis, sphincter disturbances, or erectile dysfunction.3,8,9,10
In this paper we report the case of a patient carrying the previously described p.Arg159His mutation11,12 in the cdc-40 domain, who had an unusual combination of peripheral nerve damage and lower motor neuron signs, and for whom diagnosis of IBMPFD only became clear 8 years later when he developed FTD.
Case Report
A 69-year-old Belgian Caucasian man complained of cramps in the right calf and walking difficulties that had started 6 months previous but had progressed to the point of needing support from the banisters when walking up a staircase. He did not report sensory disturbances. His personal history included arterial hypertension, hypercholesterolemia, complete deafness of the right ear due to a mastoidectomy in early childhood, and completely asymptomatic PDB that had been diagnosed 7 years previously by elevated alkaline phosphatases in a routine blood check, and had been confirmed by bone diphosphonate technetium-99m scintigraphy. His sister had died of dementia at the age of 62 years, while his mother had died at 67 years suffering from dementia and walking difficulties. At the first neurological examination he had slight paresis of the dorsal extensors of the left foot, eft quadriceps, and right deltoid muscle, atrophy of the left thigh, and diffuse tendon hyperreflexia. Fasciculations were noted in the right deltoid muscle but not in the tongue, which was not atrophic. Cranial nerves and sensibility were intact and there was no Babinski sign.
The first electromyography (EMG) investigation revealed a diffuse chronic neurogenic pattern in all tested muscles of the left leg and in the right deltoid muscle, without signs of subacute denervation such as fibrillations or positive sharp waves. There was a marked slowing of the motor nerve conduction of the right peroneal nerve, without conduction block.
An extensive laboratory workup produced normal results except for slight elevations of alkaline phosphatase (115 mU/mL, normal range 30-90 mU/mL) and lactate dehydrogenase (308 mU/mL, normal range 140-280 mU/mL). Magnetic resonance imaging (MRI) of the lumbar spine and computed tomography scans of the cervical spine and pelvis did not reveal significant abnormalities.
Walking for longer than 20 minutes had become difficult in the intervening 3 years since these initial investigations, with the patient reporting falls and intermittent paresthesias in the left big toe. The patient presented with a right Babinski sign and absent ankle reflexes.
Further electrophysiological studies (Table 1) revealed diffuse sensorimotor neuropathy with a chronic neurogenic denervation pattern in the bilateral tibialis anterior and rectus femoris muscles, the right vastus lateralis, and soleus. There were signs of subacute denervation (fibrillations and/or fasciculations) in the right tibialis anterior muscle, the left vastus lateralis, and the biceps of the right arm. The desirability of performing muscle and nerve biopsies was discussed with the patient, but they were refused.
Somatosensory evoked potentials (SEP) suggested subcortical, supramedullary slowing of central somatosensory pathways, while motor evoked potentials (MEP) induced by transcranial magnetic stimulation (TMS) of the central motor pathways revealed elevated stimulus thresholds and slightly delayed central conduction, which were restricted to the lower limbs.
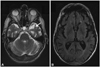
A lumbar puncture showed normal protein, glucose, cytology, and IgG index, and identical oligoclonal bands in the serum and cerebrospinal fluid. Syphilis and borrelia serology was negative. The levels of serum human T-lymphotropic virus 1 antibodies, serum arylsulphatase-A, and urinary catecholamines were normal. A further workup for polyneuropathy (including vitamin B status, diabetes screening, thyroid function, protein electrophoresis, and tumor markers) did not reveal any metabolic, inflammatory, paraneoplastic, or toxic cause of polyneuropathy. MRI of the brain showed bilateral frontotemporal atrophy (Fig. 1). MRI of the dorsal spine showed focal atrophy spanning about 5 cm of the spinal cord at the D4 level, with secondary centromedullary dilatation (unfortunately these images could not be retrieved).
The patient rapidly developed frontal lobe symptoms 8 years later. He stayed fully oriented but became apathetic, dysphoric, and verbally aggressive. He showed perseverations and paraphasias in conversation and had become deaf in his left ear. He had no hallucinations, frontal release signs, or sphincter problems, and normal oculomotricity. He had paraparesis that especially affected the psoas muscles, hamstrings, and dorsiflexors of the feet, with relative sparing of flexor muscles. There was slight muscle atrophy, which, like the paraparesis, was more pronounced in the left quadriceps (Fig. 2). However, the upper limbs could exert normal forces. The Babinski sign had disappeared but there was a bilateral Hoffmann-Trömner sign and generalized hyperreflexia, except for the ankle reflexes, which were absent. The patient refused formal neuropsychological testing.
The presence of personal and familial histories of dementia and progressive gait disturbance and the history of (albeit asymptomatic) PDB led to a putative diagnosis of IBMPFD. A sequence analysis of the 17 coding exons of the VCP gene (Born-Bünge Institute, University of Antwerp) confirmed the existence of a simple base mutation in codon 159 (Arg>His), which was confirmed in a second independent analysis. The patient died 1 year later due to a respiratory infection. An autopsy was not performed.
Discussion
To our knowledge, four families with a p.Arg159His mutation have been described. Through the screening of 123 Belgian patients with FTD, van der Zee et al.11 identified 2 families with this mutation; none of the 10 patients in these 2 families had myopathy: 6 siblings of the first family presented only FTD, with an age at onset ranging from 60 to 64 years, while 4 siblings of the second family were affected with dementia, PDB, or both, with a mean onset age of 46 years.
Haubenberger et al.12 reported four siblings of an Austrian family affected with the same mutation. The disease onset occurred between the ages of 37 and 60 years, with PDB followed by myopathy in three women, while the man had the inverse presentation. They all had proximal weakness and atrophy in the legs, which (at least in the women) later spread to the upper limbs. EMG findings were consistent with myopathy (brief and small motor-unit action potentials), which was confirmed in the male patient by muscle biopsy. This patient also had distal hypoesthesia and paresthesia of the lower limbs, and a polyneuropathy that had been attributed to diabetes. None of the patients had clinical signs of FTD.
The initial picture of the three members of the family reported by Stojkovic13 was myopathy of both upper and lower limbs. The age at onset varied from 60 to 70 years. Two had cognitive impairment, but none of them had PDB.
Since our patient refused a muscle biopsy, we cannot formally rule out that at least a part of his motor symptoms were due to unrecognized IBM. Repeated EMG explorations failed to detect features suggestive of myopathy, but this does not rule out the presence of IBM. There were clinical and electrophysiological arguments for the involvement of both the peripheral and central nervous systems: nerve conduction studies and EMG confirmed the presence of a sensorimotor neuropathy. The muscle atrophy and the absent ankle reflexes were consistent with involvement of lower motor neurons, while hyperreflexia and the results from evoked potentials implied concomitant damage to the CNS. The presence of dorsal medullar atrophy of unknown origin may have contributed to the slowed central conduction times on SEP and MEP but cannot account for the elevated stimulus thresholds on TMS.
Very few cases of IBMPFD have involved the peripheral nervous system. As mentioned above, the Austrian patient with polyneuropathy had diabetes. This was also the case in one patient with a R191Q mutation described by Spina et al.9 A review of 49 patients from 9 families revealed that 2 of the patients had diabetic neuropathy. However, a third patient from a Canadian family carrying a A232E mutation-whose clinical picture was mistaken for limb girdle muscle dystrophy and PBD-had an electrophysiological workup suggestive of axonal sensory neuropathy in the legs.4 Finally, Miller et al.10 reported two patients with pudendal neuropathy in a large British pedigree with a p.R155H mutation.
Our case report supports the hypothesis that peripheral nerve involvement is a rare feature of IBMPFD, and is in line with the findings of Kumar et al.14 who demonstrated-through elaborate electrophysiological studies of two families with IBMPFD-the presence of axonal hyperpolarization in motor and probably sensory axons, possibly via a diminished VCP-mediated breakdown of Na+/K+ pumps.
The differential diagnosis in our patient included other autosomal dominant mutations associated with FTD, especially of C9orf72 (which often has an ALS-like manifestation),5 adult polyglucosan body disease (which could have accounted for the involvement of the peripheral nervous system, ALS features, and FTD), and Nasu-Hakola disease, which can produce FTD and sensorimotor polyneuropathy. However, the last two diseases are autosomal recessive. The diagnosis of IBMPFD in our patient only became obvious after he developed FTD. Although no data were available from formal neuropsychological testing, the reported behavioral symptoms met the current clinical criteria for FTD,15 and this diagnosis was supported by the prominent atrophy of the left temporal lobe that occurred even before the clinical onset of FTD. This combined with a history of PDB (albeit asymptomatic) pointed to a VCP mutation.
This case illustrates the need to include IBMPFD in the differential diagnosis of atypical progressive disorders of gait, especially in the presence of a personal or familial history of gait disorders, PBD, or dementia. It is useful to screen patients for elevated alkaline phosphatases, and to perform a detailed radiological survey if they are elevated or when there are complaints of pain or pathological bone fractures.

 XML Download
XML Download