

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Spontaneous eye movements are useful clinical signs in coma, although they rarely have localizing value.1 The best-known exception to this rule is ocular bobbing, which consists of rapid downward conjugate movements of both eyes with a slower return to the baseline position, and is found in pontine lesions.2 Several variants of these movements have been described according to the direction and speed of the drifts.3 Ocular dipping (OD), or inverse ocular bobbing, is one of those variants. OD consists of slow downward deviation and rapid upward return of the eyes to the primary position,4 and has been described in association with hypoxic insults to the brain, although other etiologies have also been reported. Here we describe a case of OD seen in a patient presenting with progressive neurologic impairment leading to a vegetative state and death caused by Creutzfeldt-Jakob disease (CJD).
Case Report
A 66-year-old woman was admitted with a 6-week history of rapidly progressing cognitive impairment. Being previously asymptomatic, her family reported that the patient showed clumsiness with progressive inability to carry out the normal activities of daily living. Confusion combined with recent memory disturbances and abnormal involuntary movements of the left upper limb had developed in the patient. Complex visual hallucinations involving animals and plants were also present.
Her vital signs and the findings of a general examination were normal. A cognitive examination upon admission demonstrated severe impairment of short-term memory and orientation. Her speech was normal, but left visual neglect was striking. Her behavior was inappropriate due to disinhibition, with the most prominent inappropriate behavior being continuous laughter. Her pupils were in the middle position, symmetric, and reactive to light. The fundus was normal. A bedside examination of the visual fields showed no gross abnormalities, but simultagnosia was found. An examination of eye movements demonstrated the absence of smooth horizontal and vertical pursuit, and the inability to execute voluntary visually guided saccades. An en-bloc rotation of head and trunk was needed to reach a visual target. She was unable to grab an object displayed in the center of her visual field. These findings (peripheral visual inattention, optic ataxia, and acquired ocular motor apraxia) were consistent with Balint's syndrome. Oculocephalic maneuvers evoked normal reflex eye movements. Irrigating the external ears with cold water elicited normal and symmetric responses. Left-arm myoclonus, left-leg myoclonus, and continuous left-hand dystonic movements were evident during an examination. A gait examination revealed truncal ataxia. Other findings of the neurologic examination were normal.
Diffusion-weighted magnetic resonance imaging (MRI) revealed mild bilateral basal ganglia and parieto-occipital cortex hyperintensities. Electroencephalography demonstrated generalized, continuous periodic 2-Hz epileptiform discharges. Cerebrospinal fluid analysis showed a normal cell count, glucose levels, and protein levels, but a positive result for 14-3-3 protein. A presumptive clinical diagnosis of CJD was made. Genetic analysis of the prion protein gene demonstrated methionine/valine heterozygosity in codon 129 on the short arm of chromosome 20.
Over the next 2 days the patient deteriorated into cortical blindness, left hemiplegia, and thereafter stupor and coma. Myoclonic jerks now appeared continuous and generalized. Abnormal spontaneous vertical eye movements were evident from the fourth day after admission, consisting of a conjugate slow downward deflection followed by a rapid return to the primary position. These movements were intermittent and not rhythmic, and were consistent with OD. Intermixed roving eye movements sometimes appeared between OD cycles, but other abnormalities of vertical gaze were not seen. Doll's eye movements were full in all directions at every moment. The intensity and frequency of abnormal spontaneous eye movements consistent with OD progressively decreased until they disappeared 8 days after onset, before subsequently evolving into sustained downward deviation intermixed with occasional roving eye movements. The patient died 24 days after admission. An autopsy was performed after obtaining consent from relatives.
A gross examination of the brain at the autopsy revealed moderate cortical atrophy. A pathological examination demonstrated widespread spongiform changes with the presence of neuropil vacuoles, neuronal loss, and astrocytosis involving the cortical gray matter, hippocampal CA1 and CA2 sectors, amygdala, thalamus, hypothalamus, putamen, caudate, superior and inferior colliculi, substantia nigra, locus coeruleus, pontine and medullary tegmental nuclei, olivary nuclei, dentate nuclei, and molecular layer of the cerebellar cortex (Fig. 1). Diffuse synaptic and axonal deposits of prionic protein were evident after 3f4 antibody immunostaining. Scarce neuropil threads and tau-positive neurons were found in the parahippocampal gyrus and hippocampal CA2 sector. Alpha synuclein and TAR-DNA-binding protein-43 kDa staining were negative. Amyloid deposits were present within the walls of some of the small cerebral vessels. A pathologic diagnosis of sporadic CJD was made.
Discussion
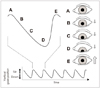
Ocular dipping is one of the spontaneous eye movements that may occur in unconscious patients.4 It is an intermittent and usually conjugate vertical eye movement consisting of a slow downward movement with a rapid return to the midpoint.5 The eyes do not elevate beyond the primary position (Fig. 2),6 and may remain in a resting downward position for several seconds before upward deviation.1 The complete cycle of movements comprising downward deviation and upward rapid recovery typically takes a few seconds. Horizontal eye movements in patients with OD usually cover the full range.6 The timing of the drift and return of the eyes to the primary position tends to vary, and the typical rhythmicity and small amplitudes of eye movements seen in nystagmus are lacking. Cycles may be continuous or intermittent; a cycle might start directly after the finish of the preceding cycle or after a short delay.7 The presence of a structural lesion involving the brainstem is very likely when other spontaneous vertical eye movements appear interspersed with OD (e.g., bobbing, reverse bobbing, or reverse dipping).5,8,9
While ocular bobbing is commonly observed in association with intrinsic pontine lesions, OD may occur in metabolic or hypoxic encephalopathies.10 OD was initially described following anoxic brain damage, but other etiologies have also been reported (Table 1). Three cases of OD in CJD have been reported that showed similar evolutions of this eye-movement disorder.11,12 OD lasted for several days in the present case before changing into downward gaze deviation. This phenomenon has been previously described in patients with OD and CJD, suggesting a complete loss of upward saccades that may be indicative of progression of the disease.11 Other disorders of involuntary eye movements in CJD include periodic alternating, upbeat, centripetal, and rebound nystagmus.11 Patients with CJD may also develop supranuclear vertical gaze palsy, slow vertical saccades, and skew deviation.4 This spectrum of eye-movement disorders reflects the prominent involvement of the cerebellum and brainstem in some patients with CJD.
The precise location of the damage leading to OD is not known. Reported cases of isolated OD (i.e., not associated with other spontaneous vertical eye movements) appearing in cerebral hypoxia have been attributed to damage to the cerebral cortex and basal ganglia on MRI or pathological investigations.10,13,14,15 The changes in intensity and frequency of OD that may occur with changes in arousal and the usually intact horizontal eye movements during vestibular-ocular testing suggest that some level of pontine function is preserved in OD and that the site responsible for its generation is located in the more rostral brainstem or in the cerebral cortex.6 Several patients have been reported with OD associated with brainstem injuries, so it has been hypothesized that structures controlling vertical eye movements are involved in the origin of OD and other variants of ocular bobbing.5,8,9,16,17,18,19 Moreover, reports of some patients showing multiple types of bobbing suggest a common underlying pathophysiology for this group of involuntary eye movements.8,20 Since different anatomical pathways mediate upward and downward eye movements, it seems likely that these movements represent varying imbalance in the mechanisms controlling vertical gaze.4
The emergence of OD usually has a poor prognosis (e.g., death, vegetative state, locked-in syndrome, or severe residual functional deficits), but some individuals have reportedly substantially improved to either complete or nearly complete recovery from their medical conditions.7,8,13,14,16 Therefore, OD cannot be used as a sign of the prognosis in coma.


 XML Download
XML Download